Key Points
- vWD is the most common hereditary bleeding diathesis, affecting up to 1% of the population; it is characterized by quantitative or qualitative deficiencies of vWF, resulting in defective platelet adhesion and aggregation.
- Type 1 vWD is the most common and the mildest form of the disease (often treated with desmopressin), while type 3 is the rarest but most severe form, requiring treatment with factor concentrates.
- Perioperative management of patients with vWD must include preoperative consultation with a hematologist (if available); the choice of perioperative drug therapy is dependent on the type of surgery, severity of vWD, baseline vWF and FVIII levels, and the patient's personal history of bleeding.
Introduction and Pathophysiology
- von Willebrand disease (vWD), first described by Finnish physician Eric Adolf von Willebrand in 1926, is the most common inherited bleeding disorder, with an estimated prevalence approaching 1% of the population.1,2
- von Willebrand factor (vWF) is a large multimeric protein involved in platelet-to-subendothelium interactions and platelet-to-platelet cohesion during thrombus formation. In addition to facilitating the binding of platelets to sites of endothelial injury, vWF also acts as a carrier for factor VIII (FVIII), slowing its clearance from plasma, and thus, increasing its plasma half-life. Normal production of vWF occurs in both vascular endothelial cells and megakaryocytes.1,3
- vWD is a heterogeneous disorder which encompasses both quantitative and qualitative deficiencies in vWF. Types 1 and 3 vWD are quantitative defects, while type 2 vWD (2A, 2B, 2M, 2N) represent qualitative defects, including disorders of platelet-to-vWF binding and vWF-to-FVIII binding (see Table 1).4
- Type 1 (mild vWD) is the most common form of vWD, with vWF levels approximately 5–30% of those in unaffected individuals. Type 2 vWD subtypes represent the second most common form. Type 3 is a rare form of vWD; however, it is also the most severe form, with extremely low (often undetectable) levels of vWF, as well as moderate deficiencies in FVIII.1,3
- Although not discussed in this summary, an acquired form of vWD (referred to as von Willebrand syndrome) has also been described in the setting of malignancy, autoimmune disease, in patients with cardiac valve abnormalities, and with certain medications.2
- In essence, vWD results from low levels of functional vWF, which in turn leads to ineffective platelet plug formation (due to quantitative/qualitative vWF defects) and poor clot stability (due to decreased FVIII levels).1
The resultant bleeding diathesis is characterized by mucocutaneous bleeding (easy bruising, menorrhagia, epistaxis) and postsurgical bleeding.1,4

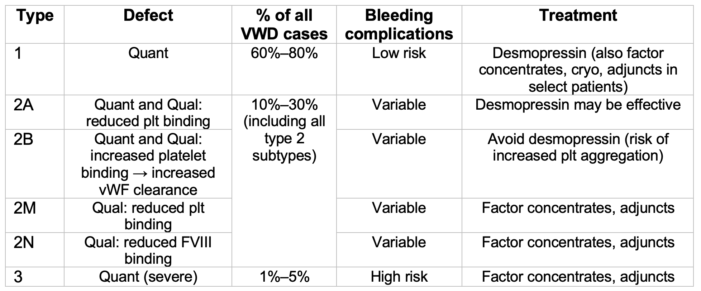
Table 1. vWD classification. Adapted from Stone ME, et al. Curr Opin Anaesthesiol. 2014.2 Qual, Qualitative; Quant, Quantitative; plt, platelet; cryo, cryoprecipitate; adjuncts include antifibrinolytics and hemostatic agents.
Diagnosis and Clinical Presentation
- Unfortunately, readily available coagulation assays such as activated partial thromboplastin time (aPTT) and prothrombin time (PT) tend to have low sensitivity and specificity for the diagnosis of vWD. For example, mild to moderate aPTT prolongation is only observed in patients with significantly-reduced FVIII levels, while platelet count and PT are often normal in patients with vWD.1,2
- The diagnosis of vWD requires an analysis of a panel of laboratory tests.1,2
- vWF antigen test (vWF:Ag): measures the concentration of vWF in the plasma
- vWF-Ristocetin Cofactor Activity test (vWF:RCo): measures the ability of vWF in the patient’s plasma to bind to normal platelets via interaction with the platelet GpIb receptor in the presence of a fixed ristocetin concentration (ristocetin is an antibiotic that stimulates vWF binding to the platelet GpIb receptor)
- FVIII coagulant assay: measures the cofactor function of FVIII
- The results of all three tests are reported in IU/dL.
- The pattern(s) of abnormal panel results, in conjunction with clinical findings, provide the diagnostic basis for specific vWD types, subtypes, and variants (see Table 2).2
- While decreased vWF:Ag is, in general, a hallmark of types 1 and 3 diseases, a decreased (less than 0.6) vWF:RCo/vWF:Ag ratio is helpful to identify type 2 disease.2
- In addition to the aforementioned laboratory assays, quantitative vWD questionnaires, bleeding assessment tools, and bleeding scores have been developed.
- One such tool is the Condensed Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD) questionnaire.1,3
- Patients with vWD classically describe a history of easy bruising, recurrent epistaxis, and menorrhagia (due to defects in platelet-mediated hemostasis).
- In patients with type 3 vWD, concomitant reductions in FVIII may lead to serious spontaneous hemorrhage, including hemarthroses.5
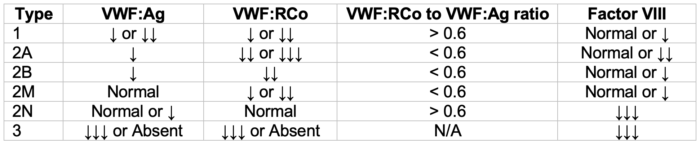
Table 2. vWD patterns of laboratory abnormalities. Adapted from Stone ME, et al. Curr Opin Anaesthesiol. 2014.2
Treatment
- Treatment strategies for vWD vary by type and severity of disease.
- Therapeutic agents used in vWD exert their effect by one or more of the following mechanisms:1,3
- Increase the level or activity of intrinsic vWF
- Replace vWF using an exogenous source
- Enhance alternative prothrombotic pathways or decrease fibrinolysis
- Examples of such therapeutic agents include:
- Desmopressin
- vWF/FVIII concentrates
- Cryoprecipitate
- Antifibrinolytic agents
- Topical hemostatics
Therapeutic Agents
- Desmopressin1
- Also referred to as 1-desamino-8-d-arginine vasopressin (DDAVP)
- Synthetic derivative of the hormone arginine vasopressin (AVP)
- Binds preferentially to the V2 vasopressin receptor subtype
- Increases the release of vWF, FVIII, and plasminogen activator from endothelial cells5
- Indications: treatment of mild type 1, mild type 2A, and type 2M vWD (effect may be too short-lived in type 2N)1,5
- Contraindicated in type 2B vWD: may increase the risk of platelet aggregation and thrombocytopenia
- Routes of administration include intravenous, intranasal, and subcutaneous.
- Tachyphylaxis often develops after repeated doses.
- Adverse effects are typically mild and include flushing, tachycardia, and headache (although rare, hyponatremia has also been reported due to desmopressin’s antidiuretic effect).
- Factor concentrates
- A variety of preparations are available, including vWF-containing FVIII concentrates, purified vWF concentrates, and recombinant vWF products.
- Humate-P is a commercially available vWF/FVIII concentrate approved for use in the United States.
- All factor concentrates are derived from human plasma are purified and then treated with virucidal techniques to decrease the risk of infection.
- Indications: prophylaxis in patients with severe type 1, severe type 2A, type 2B, type 2M, type 2N, and type 3 vWD1
- Treatment risks include possible increased DVT risk due to sustained increased levels of FVIII.1 This is due to the longer half-life of FVIII as compared to vWF in the setting of endogenous FVIII production.
- Cryoprecipitate
- Contains high levels of vWF, FVIII, FXIII, fibronectin, and fibrinogen
- Indications: in an emergency, it may be used in patients who do not respond to desmopressin or when factor concentrates are unavailable
- vWF and FVIII represent 5% of total cryoprecipitate protein.
- Cryoprecipitate is associated with higher risks of viral transmission as compared to factor concentrates.1,5
- Antifibrinolytics
- Indications: Adjunct in patients undergoing surgical procedures involving mucosal surfaces such as dental procedures (due to rich fibrinolytic activity of mucosal surfaces)1
Perioperative Management
Preoperative
- Prior to elective procedures in patients with vWD, preoperative hematology consultation should be obtained to guide perioperative management. However, such an approach may not always be feasible or practical (e.g., due to staffing constraints or in cases of emergency surgery).
- Furthermore, preoperative assessment may reveal occult vWD, e.g., in a patient with a reported family history of a bleeding disorder and a personal history of profuse bleeding from simple wounds.1 Indeed, some patients remain undiagnosed until a challenge to coagulation occurs with major surgery or antiplatelet medication exposure.
- Preoperative questionnaires, such as Bleeding Scores, have been designed to help predict postoperative bleeding outcomes.
Intraoperative
- Surgical considerations in patients with vWD are dependent on multiple factors, including the type of surgery, type of vWD, baseline vWF and FVIII levels, and the patient’s history of bleeding.3
- Current published recommendations for target levels of vWF and FVIII vary depending on the type of invasive procedure.
- It is recommended that surgeries be carried out in centers with access to laboratory testing that allow perioperative monitoring of hemostasis.
- Perioperative replacement therapy recommendations are summarized in Table 3.3
- Minor surgeries include biopsies, complicated dental extractions, gingival procedures, central line placement, or laparoscopic procedures.
- Major surgeries include craniotomy, cardiovascular, cardiothoracic, cesarean delivery, hysterectomy, or open cholecystectomy.
- Desmopressin
- Due to considerable variation in response to desmopressin among individuals, an initial test dose should be administered prior to the planned surgery, while monitoring changes in vWF and FVIII concentrations over the 4 hours following administration.5
- An intravenous dose of 0.3 μg/kg typically raises vWF concentrations by three- to four-fold.5
- Desmopressin must be given over 15 to 30 minutes in order to minimize hypotension, flushing, and tachycardia.
- Desmopressin should be used with caution in children younger than 2 years, due to the risk of hyponatremia.

Table 3. vWD surgical treatment recommendations. Adapted from Miesbach W. Eur J Haematol. 2020.3 TXA, tranexamic acid.
Postoperative
- vWF levels (vWF:RCo) should be assessed following infusion, then at least once daily following surgery.
- Plasma levels of FVIII:C should be measured every 12 hours on the day of surgery, then every 24 hours post-surgery until discharge.
References
- Mazzeffi MA, Stone ME. Perioperative management of von Willebrand disease: a review for the anesthesiologist. J Clin Anesth. 2011;23(5):418-26. PubMed
- Stone ME, et al. Current management of von Willebrand disease and von Willebrand syndrome. Curr Opin Anaesthesiol. 2014;27(3):353-8. PubMed
- Miesbach W. Perioperative management for patients with von Willebrand disease: Defining the optimal approach. Eur J Haematol. 2020;105(4):365-77. PubMed
- O’Donnell JS, Lavin M. Perioperative management of patients with von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2019;2019(1):604-9. PubMed
- Wijeysundera DN, Finlayson E. Preoperative evaluation. In: Gropper MA. Miller's Anesthesia. Ninth edition. Philadelphia, PA; Elsevier, Inc; 2020:918-98.
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.