Key Points
- Portopulmonary hypertension (PPH) is the presence of pulmonary arterial hypertension, not attributable to other etiologies, in the setting of liver disease or portal hypertension.
- The severity of liver disease is not linearly associated with the severity of PPH.
- PPH is associated with high morbidity and mortality in patients undergoing liver transplantation, necessitating the screening for PPH during the evaluation of potential liver transplant recipients.
- Preoperative optimization of the potential recipient with PPH is critically important prior to undergoing liver transplantation. Recent advances in treatment modalities have led to better outcomes.
Introduction
- PPH is present in up to 8.5% of liver transplant candidates.1
- Reports of incidence vary widely, but PPH has been reported in up to 16% of patients with severe liver disease.2
- The severity of liver disease or portal hypertension does not predict the presence or severity of PPH; however, it is more common in patients with severe liver disease.
- The World Health Organization classifies PPH in group 1, as pulmonary arterial hypertension (PAH) secondary to liver disease.3
- The most common symptoms in patients with PPH include dyspnea on exertion, fatigue, palpitations, chest pain, and syncope. This contrasts with hepatopulmonary syndrome (HPS), which typically presents with hypoxia. Please see the OA summary on hepatopulmonary syndrome for more details. Link
Pathogenesis
- Patients with portal hypertension have a hyperdynamic circulation, with high cardiac output resulting in splanchnic volume overload, bowel wall edema, and subsequent release of cytokines and endotoxins (Figure 1).

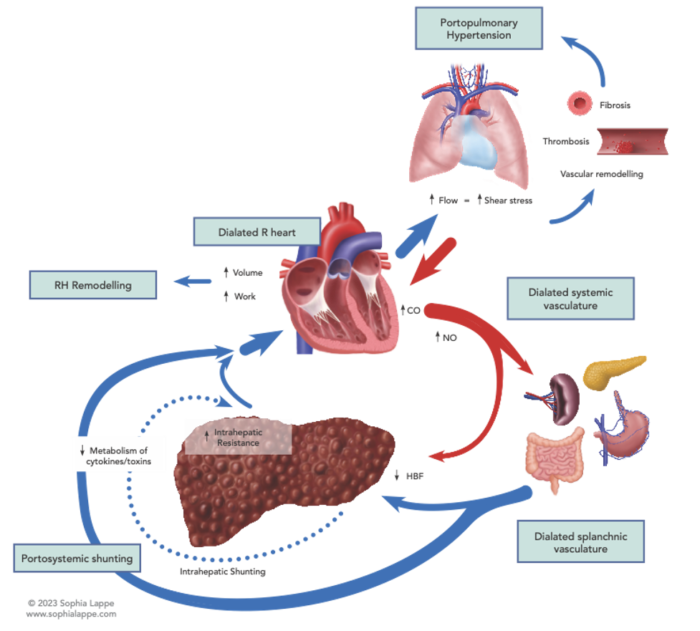
Figure 1. Factors leading to the development of portopulmonary hypertension. Abbreviations: CO, carbon monoxide; NO, nitric oxide; RH, right heart; HBF, hepatic blood flow
- This leads to increased vascular resistance and pulmonary vascular remodeling and is marked histologically by the proliferation of pulmonary endothelial cells, intimal thickening, and medial hypertrophy.4
- Portal hypertension may also lead to pulmonary vascular dilatation due to increased shear stress on a normally low resistance system, a compensatory mechanism to maintain a normal PVR. This is the hallmark of HPS, which must be differentiated from PPH in patients with cirrhosis and pulmonary symptoms (Figure 2).
- The process is further complicated by in-situ thrombosis as well as chronic pulmonary emboli, which leads to the development of “plexiform lesions” (defined by their web-like histological appearance) secondary to recurrent thrombosis, fibrosis, and recanalization. This pathology is histologically identical to those seen in primary PAH.
- Genetic factors likely contribute to the individual risk for developing PPH in the setting of severe liver disease and is a major area of ongoing research.
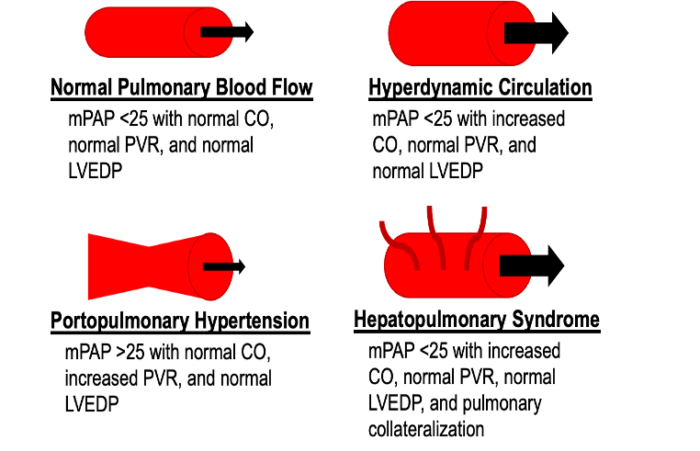
Figure 2. Differentiating PPH from hepatopulmonary syndrome and high-flow states Normal parameters: CO 4-8L/min; PVR <3 Wood units; LVEDP ≤ 15mmHg Abbreviations: mPAP, mean pulmonary artery pressure; CO, cardiac output; PVR, pulmonary vascular resistance; LVEDP, left ventricular end-diastolic pressure
Diagnosis & Management
- Diagnosis of PPH requires the presence of the following:
- Portal hypertension
- mPAP >25 mmHg at rest, >30 mmHg with exertion or PVR > 3 Wood units
- LVEDP ≤ 15mmHg
- Exclusion of other pulmonary hypertension etiologies
- Evaluation typically includes:
- Electrocardiogram
- Chest radiograph
- Pulmonary function testing
- Transthoracic echocardiogram
- Right heart catheterization (if predicted RVSP ≥ 50mmHg)
- Medical management: limited data exists for the use of the following medications in patients with PPH. However, data from patients with primary PAH have been extrapolated to guide these therapies in PPH.
- Prostacyclins: epoprostenol, treprostinil, iloprost
- Endothelin receptor agonists: bosentan, ambrisentan
- Phosphodiesterase inhibitors: sildenafil, tadalafil
- Liver transplantation:
- Multiple small case series have demonstrated that liver transplantation can stabilize, improve, or even cure PPH.
- Prior to the implementation of aggressive optimization and medical management, outcomes were very poor after liver transplantation in patients with PPH.5
Prognosis
- In liver transplant patients with severe untreated PPH, 5-year survival has been reported to be ~10%, with survival improving to nearly 50% with the addition of vasodilator therapies.6
- Median survival in patients with severe untreated PPH is ~6 months.1
- In a hallmark meta-analysis, patients with mPAP 35-40mmHg had a 50% mortality rate after liver transplantation, and patients with mPAP ≥ 50mmHg had a 100% mortality rate after liver transplantation.7
References
- Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. 2008;178(6):637-43. PubMed
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet. 2004;363(9419):1461-8. PubMed
- Rich S. Primary pulmonary hypertension: executive summary from the world symposium. WHO Programme on Cardiovascular Diseases, 1998. Available at Link Accessed March 25th, 2024.
- Budhiraja R, Hassoun PM. Portopulmonary hypertension: a tale of two circulations. Chest. 2003;123(2):562-76. PubMed
- Cartin-Ceba R, Krowka,MJ. Portopulmonary hypertension. In: Cirrhosis: A practical guide to management. Lee SS, Moreau R (eds) Wiley Blackwell, 2015.
- Safdar Z, Bartolome S, Sussman N. Portopulmonary hypertension: an update. Liver Transpl. 2012;18(8):881-91. PubMed
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6(4):443-50. PubMed
Other References
- Rubin LJ. Portopulmonary hypertension. In: Post T (ed). UpToDate. 2023. Accessed Mar 24, 2024. Link
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.