Key Points
- Neuromuscular transmission is mediated by the release of the neurotransmitter acetylcholine (ACh) and its effect on postjunctional acetylcholine receptors (AChRs). Neuromuscular blockade is achieved by disrupting this pathway.
- Depolarizing neuromuscular blocking agents, such as succinylcholine, open the AChR ion channels, depolarize the motor endplate, and prevent further neuromuscular transmission.
- Nondepolarizing neuromuscular blocking agents, such as rocuronium, competitively inhibit ACh from binding to the nAChR, preventing conformation change needed to induce depolarization.
- Brain stem reflexes are typically preserved with neuromuscular blockade.
Anatomy and Physiology of NMJ1
- The neuromuscular junction (NMJ) consists of a presynaptic motor neuron, a synaptic cleft, and a postsynaptic muscle fiber (Figure 1). Transmission of an action potential in the motor neuron occurs through sodium ion influx through membrane sodium channels. This depolarization causes the opening of the Ca2+ voltage-gated channels at the presynaptic terminal. The influx of calcium releases ACh from vesicles stored in the prejunctional terminal into the synaptic cleft.

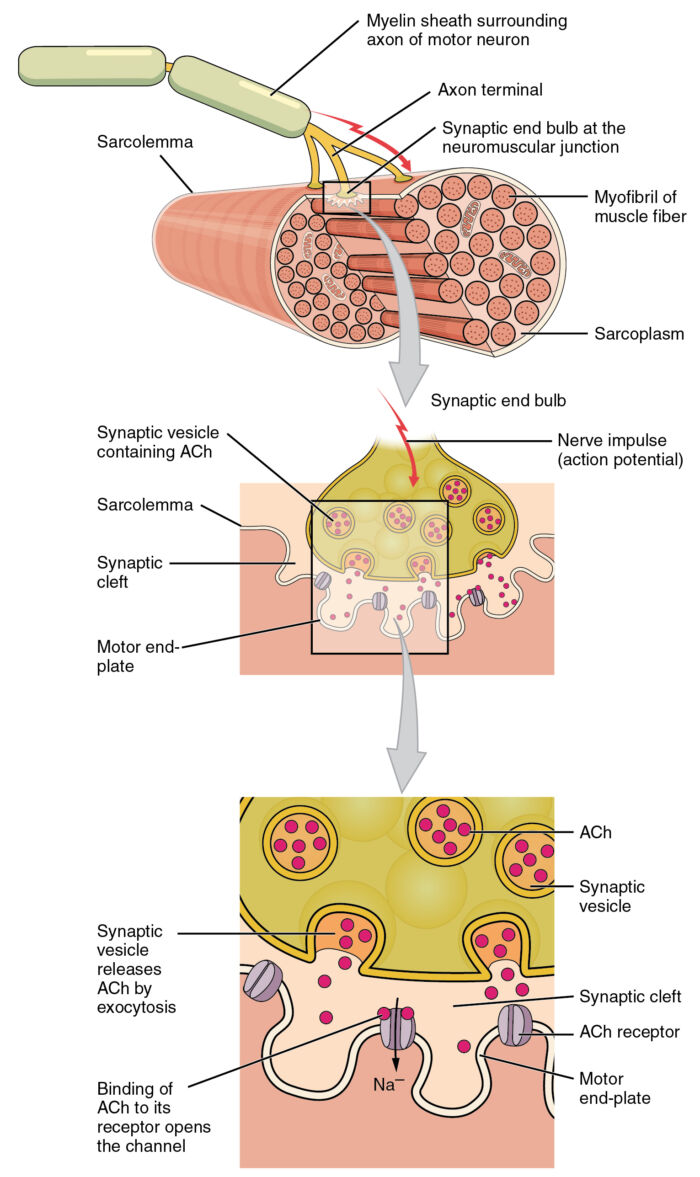
Figure 1. In the neuromuscular junction, the presynaptic motor neuron releases ACh into the synaptic cleft and binds to the receptors on the motor endplate. Adapted from Version 8.25 from the Textbook OpenStax Anatomy and Physiology (CC BY 4.0) Link.
{kind=link}
- Toxins, such as botulinum, cause muscle paralysis by blocking the release of these vesicles. ACh then binds to prejunctional and postjunctional receptors in the neuromuscular junction.
- Ligand-gated channels, known as junctional nicotinic acetylcholine receptors (nAChR), are present at the motor endplate and signal muscle contraction when bound by ACh. These receptors are composed of five protein subunits (two α, and one each of β, δ, and ε subunits), and this protein-receptor complex spans the entire membrane.
- The two α subunits of the receptor complex contain the ACh-binding sites. ACh binds to each of the two α subunits, triggering a conformational change of the ion channel, allowing the influx of Na+ ions and efflux of K+ ions, which depolarize the muscle (Figure 2). Voltage-gated Na+ channels on the muscle membrane then propagate the action potential across the membrane, inducing Ca2+ to enter the muscle cell via voltage-gated Ca2+ channels, ultimately leading to muscle contraction (excitation-contraction coupling).
- ACh is very rapidly hydrolyzed via acetylcholinesterase in the synaptic cleft. Its metabolite, choline, is then recycled back into the presynaptic nerve terminal to be synthesized and stored as ACh to start the process over again. Inhibiting acetylcholinesterase increases the lifetime of ACh in the synaptic cleft and increases the likelihood that it will bind to a receptor.
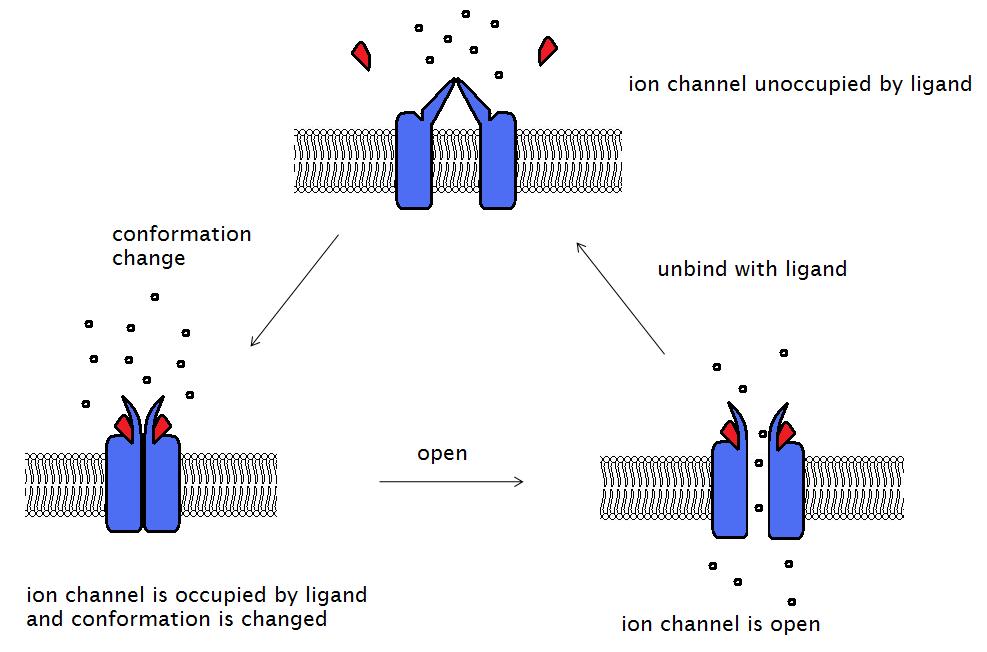
Figure 2. Binding of a ligand (e.g., ACh) to two α subunits on the AChR opens the ion channel and allows ion flow, which depolarizes the motor endplate. Source: Wikimedia, Laozhengzz. CC BY-SA 3.0. Link.
{kind=link}
Upregulation of nAChRs
- Clinically, certain disease states can cause upregulation of nAChRs as a compensatory response to decreases in the frequency of stimulation over time. These states also increase the expression of the immature, or extrajunctional isoform of the ACh receptor, in which channel opening time is up to 10-fold longer than that of mature receptors. This prolongation can result in increased sensitivity to agonists like succinylcholine. The resultant hyperkalemia can be severe enough to cause cardiac dysrhythmias.
- Receptor upregulation occurs in conditions such as immobilization, sepsis, stroke, multiple sclerosis, severe burns, denervation injury, multiple sclerosis, and prolonged use of neuromuscular blockers commonly encountered in the intensive care unit.
Neuromuscular Blocking Agents1-3
Depolarizing Neuromuscular Blocking Agents
- The structure of succinylcholine is composed of two ACh molecules linked together through the acetate methyl groups (Figure 3). Therefore, it acts as an ACh agonist at both postsynaptic and extrajunctional ACh receptors. Because succinylcholine is not metabolized immediately by acetylcholinesterase, the endplate remains depolarized. The sodium channels are inactivated, and neuromuscular transmission is blocked. This results in desensitization of the nAChR (i.e., phase I or accommodation block).
- If repeated doses or a prolonged infusion of succinylcholine is given, a phase II block can develop, which is characterized by a fade in the train-of-four response, much like a competitive block from a nondepolarizing neuromuscular blocking agent.
• Succinylcholine is hydrolyzed by plasma butyrylcholinesterase; thus, patients with pseudocholinesterase deficiency may experience a prolonged block. With atypical pseudocholinesterase, a phase II block can also develop.
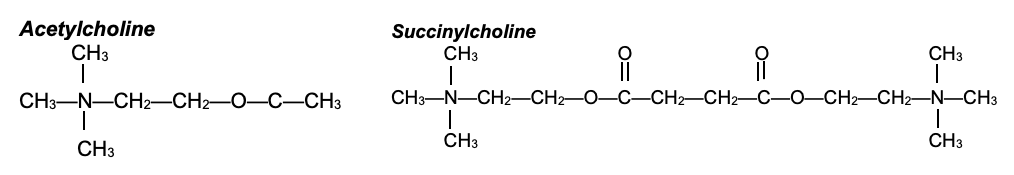
Figure 3. Structures of ACh and succinylcholine. Succinylcholine consists of two ACh molecules linked by an acetate methyl group.
Nondepolarizing Neuromuscular Blocking Agents
- Nondepolarizing neuromuscular blocking agents function as competitive ACh receptor antagonists and prevent ACh from binding to the nAChR. They are classified based on their chemical structure: steroidal (e.g., rocuronium, vecuronium, pancuronium) or benzylisoquinolinium (e.g., mivacurium, atracurium, cisatracurium).
- With an antagonistic block, the endplate potential fails to reach the threshold to initiate a propagating action potential to produce muscle contraction. This block causes muscle paralysis. Unlike depolarizing agents, nondepolarizing neuromuscular blockers do not produce a conformational change in the receptor. Because the binding of antagonists is dynamic with the repeated association and dissociation, a high enough concentration of ACh can favor ACh binding to the AChR.
NMB: Brainstem Reflexes
- The effects of neuromuscular blocking agents are typically in the periphery, but animal studies have shown that they can also exert effects centrally at neuronal nAChRs. Tubocurarine and vecuronium have been shown to depress central respiratory activity through suppression of central nAChRs in rats.4 In rabbits, pancuronium has been shown to decrease evoked electromyography but had no significant effect on auditory brainstem responses or the pupillary light reflex.5
- The preservation of the pupillary light reflex has been confirmed in the majority of humans receiving neuromuscular blocking agents.6 As described above, neuromuscular blocking agents function by either competitive blockade of ACh or persistent depolarization at the motor end plate of striated muscle. In the pupillary response, the ocular pupillary sphincter muscle is composed of smooth muscle and, therefore, may not be affected by neuromuscular blocking agents.
References
- Jeevendra Martyn JA, Fagerlund MJ. Neuromuscular physiology and pharmacology. In: Gropper M, Cohen NH, Miller RD, et al.(eds) Miller’s Anesthesia. Philadelphia, PA. 9th edition. Elsevier; 2019:333-353.
- Appiah-Ankam J, Hunter JM. Pharmacology of neuromuscular blocking agents. Continuing Education in Anaesthesia Critical Care & Pain. 2004; 4(1):2-7. Link
- Brull SJ, Meistelman C. Pharmacology of Neuromuscular Blocking Drugs. In: Gropper MA, Cohen NH, Miller RD, et al.(eds) Miller’s Anesthesia. Philadelphia, PA. 9th edition. Elsevier; 2019: 792-831.
- Sakuraba S, Kuwana S, Ochiai R, et al. Effects of neuromuscular blocking agents on central respiratory control in the isolated brainstem-spinal cord of neonatal rat. Neurosci Res. 2003; 47(3):289-98. PubMed
- Saito T, Yamamoto I, Huang XL, et al. Effects of muscle relaxants on EEG, ABR and EMG in rabbits. Hum Exp Toxicol. 1999; 18(12):718-23. PubMed
- Caro, DA, Andescavage S, Akhlaghi M, et al. Pupillary response to light is preserved in the majority of patients undergoing rapid sequence intubation. Ann Emerg Med. 2011; 57(3):234-237. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.