Key Points
- Hepatic encephalopathy (HE) is a complex neuropsychiatric syndrome associated with liver dysfunction, both in acute liver failure and chronic liver disease.
- It presents with a variety of cognitive and motor symptoms ranging from subtle behavioral impairments to coma.
- It is a common complication of end-stage liver disease (ESLD) and is responsible for considerable morbidity and repeated hospital admissions.
- Previously believed to be completely reversible after successful liver transplantation, it is now understood that some symptoms of severe HE may be permanent.
Introduction
- HE is defined as a brain dysfunction caused by liver insufficiency and/or portosystemic shunting of blood. It presents as a wide spectrum of neurological and psychiatric symptoms.1
- It arises due to the liver’s declining ability to effectively clear neurotoxic substances, especially ammonia, from the bloodstream.
- Other substances believed to contribute to HE include:
- Manganese – accumulates in the brain due to impaired hepatic excretion related to chronic liver disease.
- Aromatic amino acids (AAAs) – phenylalanine, tryptophan, and tyrosine disrupt neurotransmitter balance in the brain.
- Mercaptans and short-chain fatty acids—produced by gut flora, these compounds cross the blood-brain barrier and contribute to neurotoxicity.
- Cytokines – elevated levels of tumor necrosis factor-alpha (TNF-α) and interleukins1β and 6 in chronic liver disease are associated with heightened ammonia-induced neurotoxicity.
Pathophysiology2,3
- HE is primarily caused by impaired ammonia metabolism. Ammonia acts as a potent neurotoxin, and hyperammonemia is a central feature in the pathogenesis of HE.
- The body generates ammonia through three main processes.
- In the small intestine, gut bacteria deaminate dietary glutamine, producing ammonia as a byproduct.
- In the large intestine, colonic bacteria and mucosal enzymes metabolize dietary proteins, amino acids, and urea, generating ammonia.
- Muscle cells produce ammonia during protein metabolism, particularly through amino acid transamination and during the purine-nucleotide cycle.
- In a healthy liver, ammonia from the portal circulation is detoxified as it passes through the liver lobule. Its conversion into urea enables its excretion in the urine.
- In individuals with liver dysfunction or cirrhosis, this detoxification process is impaired due to either reduced functional liver tissue or the shunting of blood directly into the systemic circulation. This results in an accumulation of ammonia in the blood.
- Ammonia can cross the blood-brain barrier, where astrocytes absorb it and convert it into glutamine. Elevated glutamine levels cause astrocyte swelling and edema.
- Mitochondrial dysfunction triggered by elevated levels of glutamine (and ammonia from its breakdown) generates reactive oxygen species and contributes to the inflammatory oxidative stress.
- Dysregulation of the glutamate-glutamine cycle increases the brain concentration of the excitatory neurotransmitter glutamate, leading to permanent neuronal damage due to overstimulation or excitotoxicity (Figure 1).

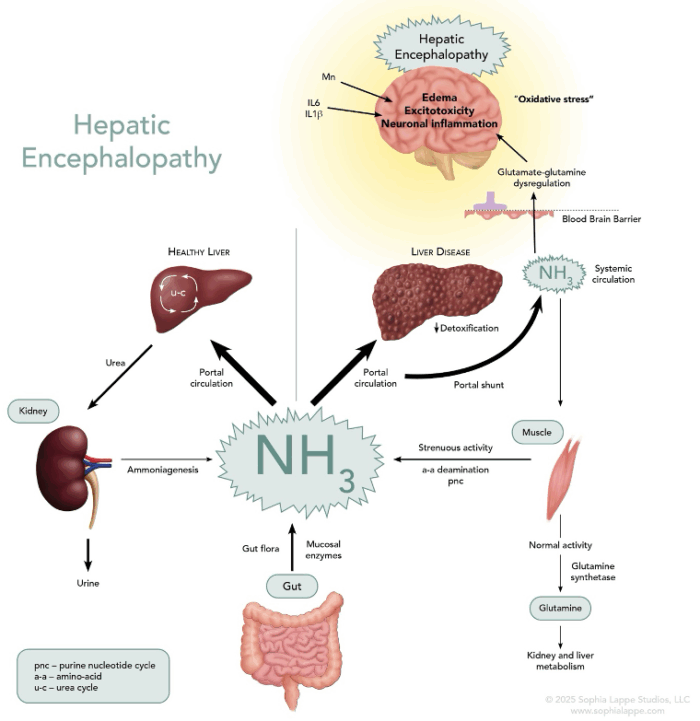
Figure 1. The pathophysiology of hepatic encephalopathy. It is thought to be primarily caused by hyperammonemia secondary to severe liver dysfunction, leading to neuronal dysfunction and cerebral edema.
Presentation
- HE can present in several ways. Often, subtle behavioral changes, such as alterations in sleep patterns, memory, psychomotor function, and concentration, are noticed first (Table 1).
- As the syndrome progresses, these changes become more overt and progress to lethargy, confusion, bizarre behaviors, and extrapyramidal motor signs (rigidity, akinesia, tremors, dyskinesia, dystonia).

Table 1. Grading of HE according to West Haven Criteria (WHC) and International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) criteria.
- Diagnosis is primarily through clinical evaluation and the exclusion of other forms of altered mental status. Hyperammonemia supports the diagnosis but is not conclusive, as other conditions can precipitate HE. Neuropsychological testing to assess cognitive and psychomotor function is recommended.5 Electroencephalography (EEG) may identify characteristic triphasic waves associated with metabolic encephalopathies, including HE.
- Computed tomography (CT) and magnetic resonance imaging (MRI) are used to rule out structural brain lesions, especially when focal signs are present.
- Predisposing factors in the development of HE include:4
- Age, severity of liver disease, and MELD score – higher values of all three indices are associated with a higher risk of developing HE.
- Infection – treatment with appropriate antibiotics should be started as soon as possible, especially for spontaneous bacterial peritonitis.
- Gastrointestinal bleeding – investigation of the cause and prompt intervention is required. Blood transfusions may be necessary.
- Hyponatremia, acute kidney injury (AKI), and dehydration – optimizing fluid status, including the aggressive management of electrolyte disturbances, can be a difficult balancing act.
- Malnutrition and sarcopenia – nutritional supplements are often required to fuel the increased energy expenditure associated with impaired digestion and muscle catabolism.
- Constipation – appropriate use of laxative medication to ensure reduced ammonia absorption.
- Sedative medications – avoiding nervous system depressants, which exacerbate the signs and symptoms of HE.
- The onset of HE reflects worsening liver disease and a poor prognosis. Patients with HE often require more health care interventions and hospital admissions, and as their quality of life deteriorates, they rapidly become a priority for liver transplantation.5
Management
- The primary aim of treatment is to reduce the ammonia loading of the brain. Limiting ammonia production and absorption in the gut is commonly achieved using lactulose and rifaximin.6
- Lactulose is a synthetic sugar that is broken down in the colon into low-molecular-weight acids that trap ammonia as the water-soluble ammonium ion in the stool. It also acts as an osmotic laxative and speeds up the transit time, thereby reducing the amount of ammonia formed by the gut flora and absorbed into the blood.
- Rifaximin is a non-absorbable, broad-spectrum antibiotic and stays in the gut. It alters the gut flora and reduces ammonia production. It is commonly used in combination with lactulose.
- L-ornithine L-aspartate (LOLA) provides substrates for the urea cycle and improves ammonia detoxification by increasing hepatocyte urea production.
- Alternative and adjunct treatments for reducing ammonia levels include the use of polyethylene glycol (PEG) as an alternative to lactulose, other antibiotics (i.e., neomycin, metronidazole, and vancomycin), dietary probiotics, and zinc supplementation.6,7
Anesthesia Management for Patients with HE
- Patients with HE often need sedation for endoscopic and other minor procedures such as placement of ports, lines, drains, colonoscopies, and endoscopies.
- The plan for anesthesia should be to minimize the risk of exacerbating HE.8
- Short-acting medications such as propofol, remifentanil, and dexmedetomidine are useful first-line drugs for sedation or general anesthesia.
- Where possible, opioids and benzodiazepines should be avoided.
- Maintaining hemodynamic parameters to support cerebral perfusion is critical.
- Invasive monitoring with arterial and central venous lines for closer management of volume and metabolic status should be considered.
References
- Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. J Hepatol. 2014;61(3):642-59. PubMed
- Rose CF, Amodio P, Bajaj JS, et al. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J Hepatol. 2020;73(6):1526-47. PubMed
- Wijdicks EF. Hepatic encephalopathy. N Engl J Med. 2016;375(17):1660-70. PubMed
- Pantham G, Post A, Venkat D, Einstadter D, Mullen KD. A new look at precipitants of overt hepatic encephalopathy in cirrhosis. Dig Dis Sci. 2017;62(8):2166-73. PubMed
- Hirode G, Vittinghoff E, Wong RJ. Increasing burden of hepatic encephalopathy among hospitalized adults: An analysis of the 2010-2014 national inpatient sample. Dig Dis Sci. 2019;64(6):1448-57. PubMed
- Phongsamran PV, Kim JW, Cupo Abbott J, Rosenblatt A. Pharmacotherapy for hepatic encephalopathy. Drugs. 2010;70(9):1131-48. PubMed
- Karvellas CJ, Bajaj JS, Kamath PS, et al. AASLD practice guidance on acute-on-chronic liver failure and the management of critically ill patients with cirrhosis. Hepatology. 2024;79(6):1463-1502. PubMed
- Bajaj JS, O'Leary JG, Lai JC, et al. Acute-on-chronic liver failure clinical guidelines. Am J Gastroenterol. 2022;117(2):225-52. PubMed
Other References
- Wagener G. Liver transplantation: Anesthetic management. In: Post T (ed). UpToDate. 2025. Link
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.