Key Points
- DiGeorge syndrome (DGS) is caused by a microdeletion of chromosome 22q11.2 and is characterized by conotruncal cardiac defects, craniofacial abnormalities, hypocalcemia, and thymic hypoplasia.
- Anesthetic management of DGS requires careful evaluation of cardiovascular anatomy, airway abnormalities, increased susceptibility to infection, and calcium homeostasis.
- Only irradiated blood products should be administered to prevent graft-versus-host disease.
Introduction
- DGS, also known as 22q11.2 deletion syndrome, results from a heterozygous microdeletion on chromosome 22q11.2.
- The classic triad of features comprises conotruncal cardiac defects, thymic hypoplasia leading to cellular immunodeficiency, and hypocalcemia secondary to the absence of the parathyroid glands.
- Other features include seizures, craniofacial abnormalities, palatal abnormalities, feeding difficulties, congenital gastrointestinal anomalies, behavioral problems, and developmental delay. The variability of these phenotypes may pose challenges in perioperative management.
- DGS is also known as velocardiofacial syndrome and is referred to as CATCH-22, a mnemonic for Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, and Hypocalcemia
Etiology
- As Dr. Angelo DiGeorge first described in 1968, DGS results from the loss of multiple genes on chromosome 22q11.2, with the TBX1 gene being a major part of its pathogenesis.1
- TBX1 plays a vital role in embryonic development, particularly in the formation of the pharyngeal arches, which give rise to the head, face, oropharynx, heart, great vessels, parathyroid glands, and thymus.2 The disruption of these developmental processes gives rise to the characteristic features of DGS.
- The broad feature variability is due to the size of the deletion and the involvement of additional genes in the affected region.2 This microdeletion occurs in about 1 in 4,000 to 6,000 live births.1,3 While most cases arise spontaneously, 6–10% are inherited from parents revealing minimal symptoms.4
- Routine testing for DGS in newborns was introduced in the late 1990s, replacing earlier, less reliable diagnostic methods. Advances in medical care and surgical techniques have significantly improved survival outcomes for patients with DGS.3
Pathophysiology

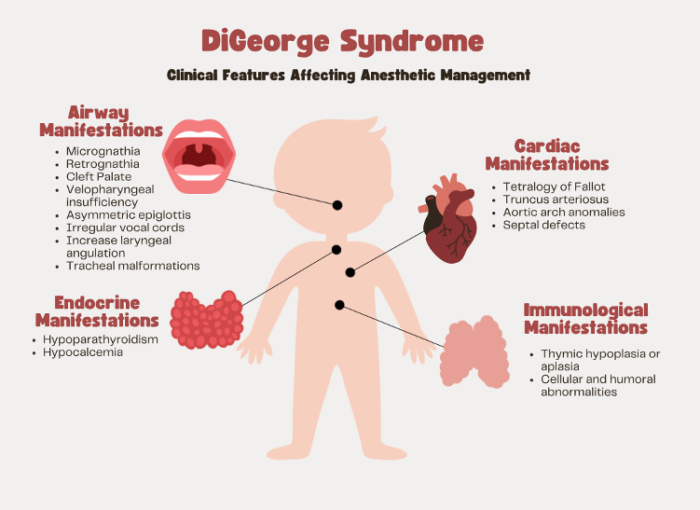
Figure 1. Clinical features of DiGeorge syndrome
Conotruncal Heart Defects
- About 75% of patients with DGS have congenital heart defects, with the majority of these being conotruncal in origin.3
- The most common cardiac anomalies include tetralogy of Fallot, truncus arteriosus, interrupted aortic arch, ventricular septal defects, and atrial septal defects. Additionally, anomalies of the aortic arch, such as right-sided aortic arches or aberrant subclavian arteries, frequently occur with other defects.3,4
- These anatomical defects have several clinical implications, including changed hemodynamic patterns, increased risk of heart failure, and pulmonary hypertension. The severity of the physiologic impact due to the anatomical defect may necessitate early surgical intervention.
- The presentation and severity of congenital heart defects may vary according to the age at diagnosis; fetuses, neonates, and older children will present differently depending upon the timing and method of detection.2,3
Airway Defects
- DGS is associated with numerous craniofacial and airway anomalies.
- Common craniofacial features, such as micrognathia, retrognathia, cleft palate, and velopharyngeal insufficiency, can alter the airway anatomy, making direct laryngoscopy or supraglottic airway placement difficult or sometimes impossible.1,2,5
- Other airway anomalies, such as asymmetric epiglottis, irregular vocal cords, increased laryngeal angulation, and tracheal malformations, may further complicate airway management.4
Immune Defects
- Most patients with DGS have some degree of immunodeficiency, primarily due to thymic hypoplasia, which leads to reduced T-cell production and impaired cellular immunity.
- Although true thymic aplasia with complete absence of T cells is rare, most patients have reduced peripheral T-cell counts, which predisposes them to recurrent and serious infections.5
- Immune function often improves with age, generally by two years, because extrathymic sources may contribute to T-cell production.2
- In addition to T-cell deficiencies, about 10% of the patients also have immunoglobulin deficiencies, with reduced levels of IgG, IgM, or IgA.5
- The combination of cellular and humoral immune dysfunction puts patients at risk for opportunistic infections and complications, such as graft-versus-host disease following transfusions.5
Endocrine Defects
- Hypoparathyroidism is the primary endocrine manifestation due to underdeveloped parathyroid glands, which results in hypocalcemia.5
- Hypocalcemia may be seen in up to 60% of neonates and can present with jitteriness, tetany, or seizures.2
- Parathyroid function may improve during the first year of life as existing parathyroid tissue hypertrophies.5
- Latent hypoparathyroidism can be seen, and episodes of hypocalcemia can be provoked by respiratory alkalosis or rapid blood transfusion, where calcium binds to citrate.4,5
- Persistent hypocalcemia requires careful monitoring and treatment with calcium and vitamin D supplementation.
- Frequent checks of serum ionized calcium levels are essential, particularly during periods of stress, to prevent complications such as hemodynamic instability, seizures, and cardiac arrhythmias.4
Anesthetic Considerations
- Administering anesthesia to patients with DGS requires careful preparation due to the complex systemic and congenital abnormalities.
- A thorough preoperative assessment is essential, including a review of:
- Abnormalities in cardiovascular anatomy and previous interventions
- Airway abnormalities/airway assessment
- Increased susceptibility to infection
- Calcium homeostasis
- Furthermore, DGS patients are at increased risk of postoperative complications, such as prolonged intubation, immune-related infections, extended intensive care stays, and increased use of hospital resources.2,3
Cardiac Considerations
- Intraoperative strategies must be tailored to the patient’s specific anatomical and hemodynamic conditions.
- Management is influenced by whether their congenital cardiac malformations have been corrected.
- Before cardiac repair, anesthesia management must balance oxygenation, pulmonary and systemic vascular resistance to prevent shunting, and myocardial function to prevent hemodynamic instability.1,4
- After cardiac repair, anesthetic considerations focus on managing residual cardiac dysfunction, pulmonary hypertension, and fluid optimization while minimizing myocardial depression.4
- For many conotruncal defects, as well as interrupted aortic arch, maintaining the patency of the ductus arteriosus is vital to ensure adequate perfusion.4 Continuous monitoring of cerebral and systemic perfusion in both the upper and lower body by using arterial catheters and pulse oximetry is essential.3,4
Pulmonary Considerations
- Airway and respiratory considerations are critical in managing anesthesia for DGS patients due to their predisposition to airway abnormalities and respiratory complications.
- Craniofacial anomalies such as cleft palate, micrognathia, retrognathia, and structural airway issues, including laryngomalacia, tracheomalacia, and subglottic narrowing, may complicate intubation.2,4
- While most intubations are straightforward, accessibility to backup equipment such as video laryngoscopy or fiberoptic bronchoscopy is advisable.
- Endotracheal tube depth may need to be modified secondary to shortened tracheal anatomy to avoid right mainstem intubation.2
- Predisposition to gastroesophageal reflux disease and pharyngeal dysmotility may increase aspiration risk.2
- Preoperative chest imaging can assist in identifying anatomical variations and guide the selection of airway devices.
- For cases requiring lung isolation, smaller-diameter double-lumen tubes or single-lumen tubes with endobronchial blockers may minimize the risk of airway injury.4
- Given the potential for prolonged postoperative ventilation, reintubation, or even tracheostomy, a thorough preoperative airway evaluation is essential to anticipate and manage these challenges effectively.
Immunological Considerations
- Immunologic considerations are crucial in DGS patients’ anesthetic plan due to common immunodeficiencies and increased susceptibility to infections.
- Preoperative assessment of T-cell function, flow cytometry, and immunoglobulin levels are essential to guide transfusion and perioperative care.5
- For patients with known or suspected immunodeficiency, only irradiated blood products should be used to prevent transfusion-associated graft-versus-host disease, and cytomegalovirus-negative or leukocyte-depleted components are recommended.3,5
- Immunodeficient patients may also require the continuation of prophylactic antibiotic regimens during the perioperative period alongside standard pre-incision prophylactic antibiotics.4
- Depending on the degree of immunodeficiency, additional antifungal or antiviral coverage may be necessary.4
- These precautions are vital, as DGS patients are at increased risk for severe perioperative infections, including sepsis, pneumonia, and endocarditis.
Endocrine Considerations
- Endocrine considerations, specifically calcium monitoring, are important for anesthetic planning in patients with DGS.
- Hypocalcemia, commonly associated with subclinical hypoparathyroidism, can lead to hemodynamic instability, seizures, bradycardia, heart failure, and arrhythmias such as QT prolongation or torsades de pointes.4
- These risks are heightened during periods of physiologic stress, such as surgery, when reduced parathyroid hormone reserves unmask the underlying hypoparathyroidism.
- To prevent complications, ionized calcium levels should be closely monitored during the perioperative period.5
- Avoid exacerbating factors of hypocalcemia, such as respiratory alkalosis from hyperventilation and citrate chelation during blood transfusion.4,5
- Treatment typically involves calcium (acute) and vitamin D supplementation (chronic), which will help maintain stable calcium levels and reduce adverse outcomes.2
References
- Yotsui-Tsuchimochi H, Higa K, Matsunaga M, et al. Anesthetic management of a child with chromosome 22q11 deletion syndrome. Pediatric Anesthesia. 2006;16(4):454-7. PubMed
- Yeng Yeoh T, Scavonetto F, Hamlin RJ, et al. Perioperative management of patients with DiGeorge syndrome undergoing cardiac surgery. Journal of Cardiothoracic and Vascular Anesthesia. 2014;28(4):983-9. PubMed
- Altshuler E, Saidi A, Budd J. DiGeorge syndrome: consider the diagnosis. BMJ Case Rep. 2022;15(2): e245164. PubMed
- Harrison JH, Dhawan R, Essandoh MK, et al. Complex reoperation in a patient with DiGeorge syndrome. J Cardiothorac Vasc Anesth. 2020;34(6):1655-62. PubMed
- Goldmuntz E. 22q11.2 deletion syndrome and congenital heart disease. Am J Med Genet C Semin Med Genet. 2020;184(1):64-72. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.