Key Points
- Cystic fibrosis (CF) is a chronic, progressive, multisystem disease that primarily affects the lungs, pancreas, hepatobiliary and gastrointestinal systems, and reproductive organs.1,2
- CF is caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, leading to impaired transport of sodium, chloride, and bicarbonate across epithelial surfaces.1,2
- Key anesthetic considerations include providing airway clearance therapies (ACTs) and inhaled airway clearance agents the morning of surgery, using warm, humidified gasses and bronchodilators, multimodal analgesia, and avoidance of atelectasis.1
Pathophysiology and Diagnosis
- CF is the most common, life-shortening, autosomal recessive disease among Caucasian populations, with an incidence of 1 in 3200 births among Caucasians in the United States.1
- CF is caused by mutations in a gene on chromosome 7 that encodes the CFTR protein.1
- The CFTR protein transports chloride and bicarbonate and impacts sodium transport across the plasma membrane of epithelial cells throughout the body, including the airways, intestines, liver, reproductive organs, and sweat glands (Figure 1).1

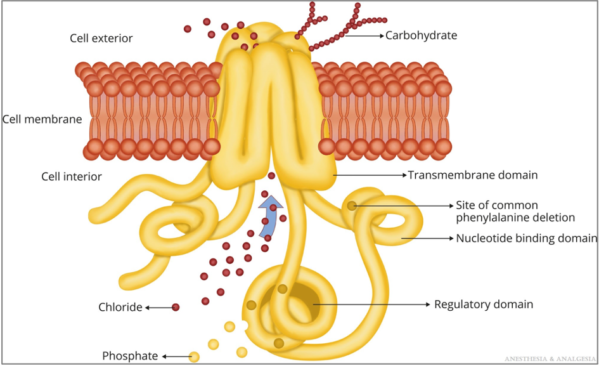
Figure 1. CFTR protein. Reproduced with permission from Lee A, et al. Anesth Analg. 2022.1
- CFTR mutations are grouped into 6 functional classes based on the primary molecular defect. (Figure 2) F508del is the most common mutation and is caused by the deletion of 3 nucleotides encoding phenylalanine (abbreviated as F) at position 508 in the CFTR protein.1
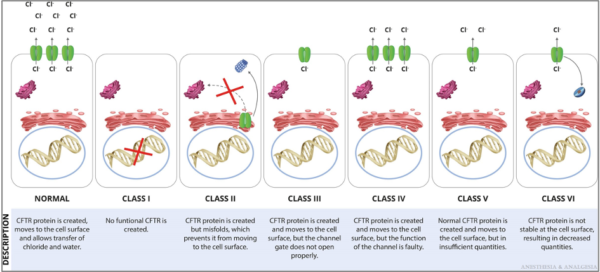
Figure 2. Functional classification of CFTR mutations. Reproduced with permission from Lee A, et al. Anesth Analg. 2022.1
- Newborn screening for CF has been widely implemented in the United States. The diagnosis of CF is based on clinical findings, sweat chloride testing, CFTR gene sequencing, and measurements of nasal potential differences (Figure 3).1
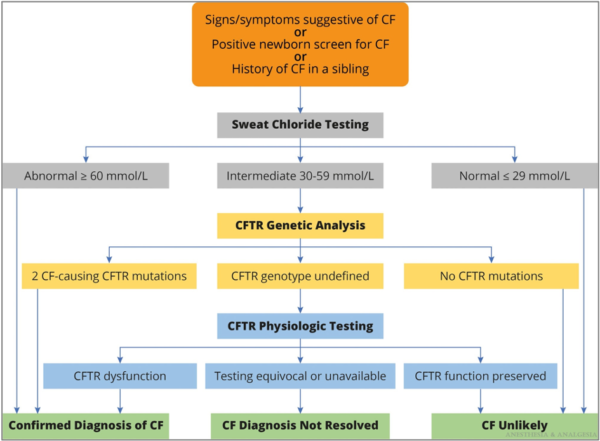
Figure 3. Algorithm for the diagnosis of CF. Reproduced with permission from Lee A, et al. Anesth Analg. 2022.1
- Significant advances in diagnosis and management have changed the epidemiological profile of CF with dramatic improvements in survival. Currently, there are more adults than children with CF in the United States and the median predicted survival age is 59 years.1-3
Clinical Manifestations and Treatment
- CF is a multisystem disease that primarily affects the lungs, pancreas, hepatobiliary, gastrointestinal systems, and reproductive organs.1
Pulmonary Disease
- Progressive lung disease is the major cause of morbidity and mortality in CF patients.
- Abnormal chloride, sodium, and water transport across respiratory epithelium leads to thick secretions, decreased mucociliary clearance, airway obstruction, inflammation, bronchiectasis, and colonization with Haemophilus influenzae, methicillin-resistant Staphylococcus aureus, Pseudomonas aeruginosa, and other bacteria, fungi, and viruses (Figure 4).1
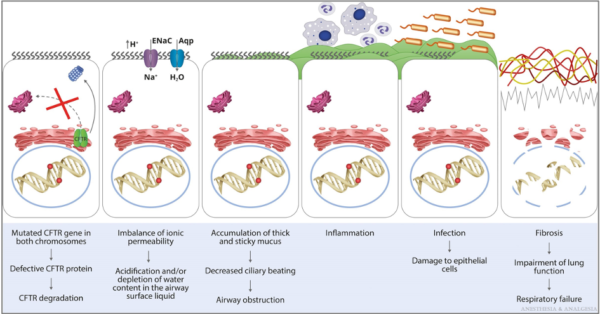
Figure 4. Pathophysiology of the pulmonary manifestations of CF. Aqp, aquaporin; ENaC, epithelial sodium channel. Reproduced with permission from Lee A, et al. Anesth Analg. 2022.1
- Respiratory symptoms include chronic rhinosinusitis, persistent productive cough, wheezing, tachypnea, dyspnea, and pulmonary hypertension in advanced disease.
- Pulmonary function tests reveal an obstructive pattern with increased residual volume (RV) and total lung capacity (TLC) and decreased forced expiratory flow at 25% to 75% of lung volume (FEF25–75), forced expiratory volume in one second (FEV1), and the ratio of FEV1 to forced vital capacity.1-3
- Treatment of pulmonary symptoms includes ACTs (breathing/coughing maneuvers, oscillating positive expiratory pressure devices, percussive vests, and high-frequency chest compressions), and inhaled airway clearance agents (dornase alfa, hypertonic saline, mannitol, beta-2 receptor agonists).1
- Oral azithromycin, inhaled tobramycin, and inhaled aztreonam lysine are recommended in all CF patients with P. aeruginosa infections.1
Pancreatic Disease
- About 85% of CF patients develop pancreatic insufficiency (PI) before 1 year of age.1
- Thick secretions block the lumen of pancreatic ducts and interfere with the secretion of pancreatic enzymes, resulting in exocrine PI, weight loss, steatorrhea, and malabsorption of fat and protein.
- CF-related diabetes (CFRD) results from the destruction of pancreatic islets, which leads to a relative insulin deficiency.
- Treatments include pancreatic enzyme replacement therapy, fat-soluble vitamin supplementation, and insulin therapy for CFRD.
Hepatobiliary Disease
- CF-related liver disease results from biliary obstruction, periportal fibrosis, and hepatic steatosis, leading to hepatomegaly, elevations in liver enzymes, cholecystitis, portal hypertension, and cirrhosis.1
- Treatment options include ursodeoxycholic acid cholecystectomy for gall bladder issues.
Gastrointestinal Disease
- About 10% of neonates with CF present with meconium ileus and small bowel obstruction.1
- Adults with CF suffer from symptomatic gastroesophageal reflux and distal intestinal obstruction syndrome, which is characterized by acute episodes of partial or complete obstruction of the terminal ileum and proximal colon by the inspissated intestine.
Genitourinary Manifestations
- About 98% of men with CF become infertile due to obstructive azoospermia.1
- Fertility in women is affected by malnutrition, thick and tenacious cervical mucus, delayed menarche, and ovulation abnormalities. About 50% of women with CF are able to conceive, and pregnancy should be carefully planned and managed by a multidisciplinary team.
Other Manifestations
- Additional complications include low bone mineral density, thrombophilia, iron-deficiency anemia, nephrolithiasis, and nephrocalcinosis.
CFTR Modulator Therapies
- Novel CFTR modulator drugs improve the production, intracellular processing, and function of the defective CFTR protein. CFTR modulators are classified into 5 main groups based on their mechanism of action.1
- CFTR modulators improve lung function and respiratory-related quality of life, increase body mass index, and reduce acute pulmonary exacerbations.1,2
- Currently, four CFTR modulators are being used in CF patients with eligible CFTR gene mutations (Table 1).1
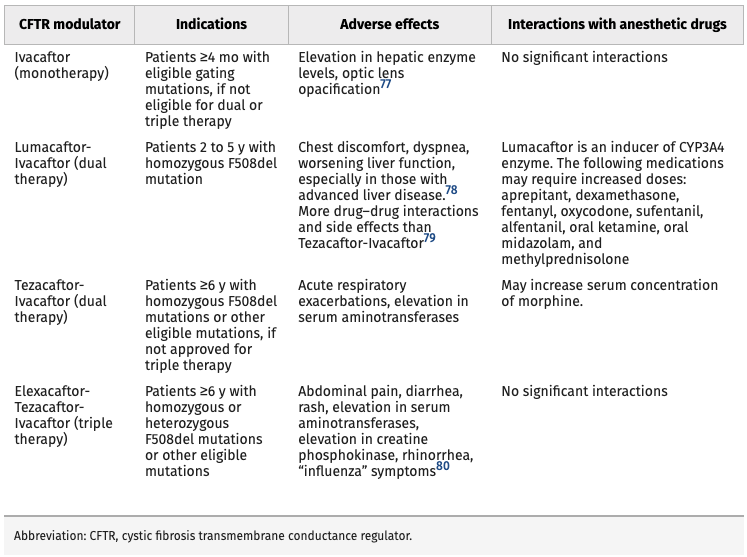
Table 1. Current CFTR Modulator Therapies. Reproduced with permission from Lee A, et al. Anesth Analg. 2022.1
Solid Organ Transplantation
- Additionally, solid organ transplantation, including double-lung transplantation and liver transplantation is a viable treatment option for CF patients with end-stage disease.
Perioperative Anesthetic Considerations
- Patients with CF are scheduled for various surgical and medical procedures. Neonates may require a laparotomy or laparoscopy for meconium peritonitis or intestinal atresia. Children and adolescents are frequently scheduled for nasal polypectomy, central venous access, or bronchoscopy and lavage. Adults commonly present for cholecystectomy, endoscopic retrograde cholangiopancreatography, endoscopy for esophageal varices, chest tubes for recurrent pneumothorax, or obstetric care. Patients with advanced disease may undergo lung or liver transplantation.1,2
Preoperative Assessment1,2
- Complete history and physical, focusing on CF-related comorbidities
- Presence of cough; quantity and quality of mucus production
- Frequency of respiratory infections
- Airway reactivity, wheezing, and response to bronchodilators
- Exercise and functional capacity
- Review chest radiographs, chest computed tomography, pulmonary function tests, baseline arterial blood gas and laboratory investigations
- Schedule procedure later in the day to allow for scheduled airway clearance therapies and inhaled airway clearance agents
- Consider delaying elective procedures by 4-6 weeks following a pulmonary exacerbation
Intraoperative Management1,2
- Routine American Society of Anesthesiologists monitors (invasive monitors if indicated)
- Common goals: minimize respiratory depression, full recovery from neuromuscular blockade at the end of the case, and early extubation to decrease morbidity and mortality.
- Avoid nasopharyngeal airways due to sinonasal disease.
- Humidify and warm airway gasses to avoid further inspissation of secretions.
- Use bronchodilators for reactive airways.
- Propofol is effective at blunting airway reflexes and is commonly used for induction and maintenance of general anesthesia.
- Volatile agents are potent bronchodilators, and are also used as a part of the anesthetic. However, desflurane should be avoided as it increases airway reactivity.
- Consider adjunctive pain management medications and techniques including regional anesthesia.
- Prior to extubation, consider chest physiotherapy, lung recruitment maneuvers, and endotracheal suction to mobilize secretions and prevent atelectasis.
Postoperative Management1,2
- Multimodal analgesia to decrease opioid requirement.
- Early ambulation, continue scheduled chest physiotherapy, inhaled airway clearance, and bronchodilators.
- Close monitoring of patients who require home supplemental oxygen or noninvasive ventilation.
- Consider overnight hospitalization for monitoring in patients with moderate to severe pulmonary disease.
References
- Lee A, Huffmyer J, Thiele E, et al. The Changing Face of Cystic Fibrosis: An Update for Anesthesiologists. Anesthesia and Analgesia. 2022; 134(6):1245-1259. PubMed
- Williamson D, Sharma A. Cystic Fibrosis in Children: A Pediatric Anesthesiologist’s Perspective. Pediatric Anesthesia. 2022; 32(2):167-173. PubMed
Other References
- Cystic Fibrosis Foundation 2020 Patient Registry Annual Data Report. Accessed May 21, 2022. Link
- Chatterjee D. Cystic fibrosis update. OpenAnesthesia. Published October 1, 2020. Accessed February 6, 2023. Link
- Chatterjee D. Interview with Dr. Pamela Zeitlin on the topic of cystic fibrosis. OA-SPA Ask the Expert podcast. Published October 1, 2020. Accessed February 6, 2023. Link
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.