Key Points
- Coagulation is a complex physiologic process balancing prothrombotic and antithrombotic factors.
- Improved understanding of the coagulation pathway in vivo has reclassified the classic coagulation cascade into three stages: initiation, amplification, and propagation.
- Fibrinolysis is a parallel system activated alongside the coagulation pathway to limit the size of clot formation.
Introduction
- The concept of blood coagulation originated in 1960s when the “waterfall model” of coagulation cascade was first used to describe the cascade of proenzymes activating downstream enzymes.

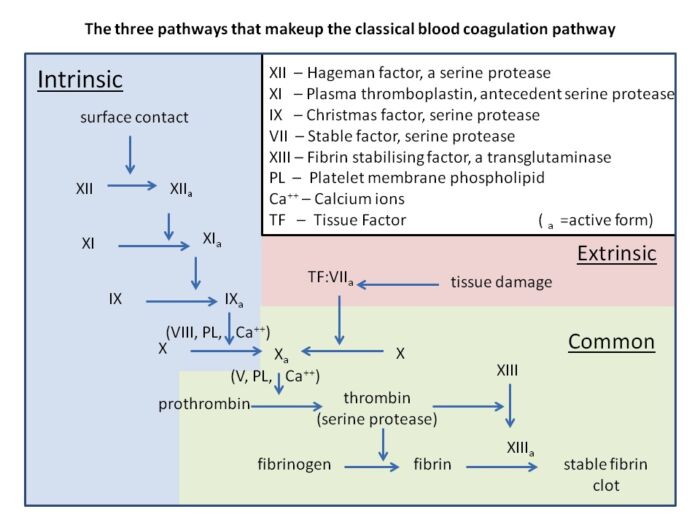
Figure 1. Cascade model of fibrin formation. This model divides the coagulation system into separate redundant pathways (extrinsic and intrinsic), either of which can result in the generation of FXa. Source: Wikipedia: Dr. Graham Beards. CC BY SA 3.0
- Multiple thrombogenic and antithrombogenic components regulate clot formation and maintain homeostasis within the coagulation system.
- In the perioperative setting, the balance of the coagulation system is often disturbed by factors such as trauma, infection, cytokines, and hypothermia.
- Coagulopathies can be broadly classified as disorders that affect primary hemostasis, coagulation pathway, and fibrinolytic system.
Coagulation Pathway
- Traditionally, the coagulation pathway has been classified into intrinsic and extrinsic pathways that converge on factor Xa activation.
- This classical theory of coagulation is useful for understanding in vitro coagulation but fails to explain certain aspects of in vivo coagulation. One of the primary deficiencies of this model is its lack of explanation for why patients with Factor XII (FXII) deficiency have prolonged intrinsic laboratory abnormalities but no clinical bleeding tendencies.1
- Current evidence supports a cell-based model of coagulation in vivo, which describes the process in three main stages: initiation, amplification, and propagation (Figure 2).
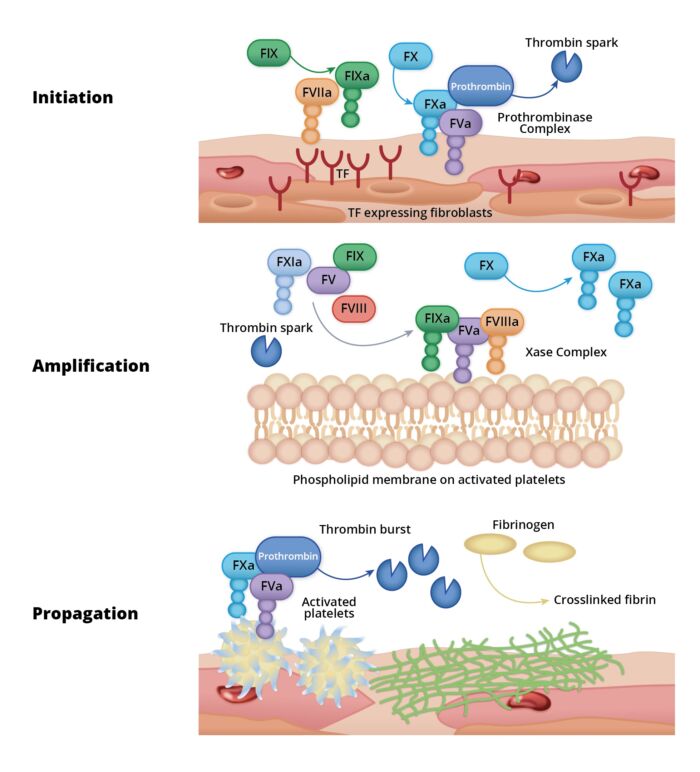
Figure 2. Cell-based coagulation model. Redrawn from O'Donnell J, et al. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br J Haematol. 2019;186(1):24-36.1
Initiation
- Tissue factor (TF) expressed on fibroblasts binds to activated FVII (FVIIa).
- FVII activation is poorly elucidated, but it is postulated to be a limited proteolytic process.
- TF-VIIa enzymatic complexes activate factor IX (FIXa) and factor X (FXa), which in turn converts prothrombin (FII) to thrombin (FIIa). This is known as the thrombin ignition phase.
- Tissue factor pathway inhibitor (TFPI) rapidly neutralizes the TF-VIIa and FXa in the absence of endothelial damage, thereby preventing the progression of the coagulation cascade.2
- Clinical Pearls
- Current evidence suggests that roughly 1% of FVII is circulating in free-activated form. This free form has very limited enzymatic activity; however, when bound to TF, it catalyzes the conversion of IX to IXa very efficiently. Factor IXa has a significant role in maintaining this free form of FVIIa process as patients with hemophilia B have much reduced free FVIIa levels.
Amplification
- If subendothelial structures are exposed, there is platelet aggregation and FIIa binds to platelets via glycoprotein Ib receptors, resulting in their full activation. Factor IIa will further activate factor XI (FXIa), V (FVa) and VIII (FVIIIa) (Figure 3).1,2
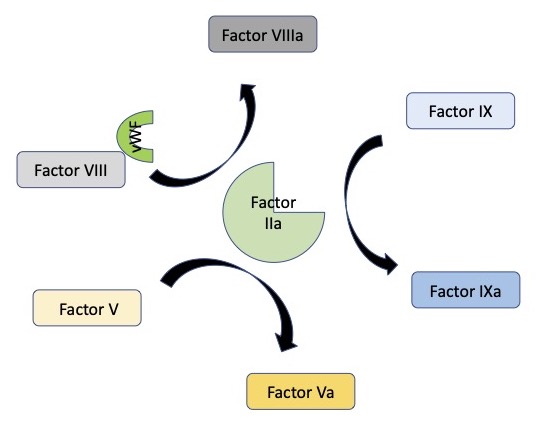
Figure 3. Amplification stage where factor IIa plays a critical role in the activation of factors V, VIII, and IX.
- As more activated platelets accumulate at the site of injury, their phospholipid layer becomes the critical site of all enzymatic processes, where XIa catalyzes the production IXa and Xa.
- Clinical Pearls
- FV: It has both procoagulant and anticoagulant properties. It is present in high levels in platelets and is released during platelet activation. FV is a coenzyme involved in thrombin ignition. It is activated by IIa and Xa (lesser extent) in its procoagulant function. In its anticoagulant role, it enhances the activity of TFPI and, along with Protein S as a co-enzyme, mediates VIIIa proteolysis by activated protein C (APC).
- FVIII: It is released from the endothelium; therefore, its concentration is not affected in trauma. In some instances, it is even increased as it is released from healthy endothelial cells in response to stress hormones (epinephrine, vasopressin, bradykinins). Thrombin ignition is typically affected if FVIII levels fall less than 20%.
Propagation
- Accumulated enzyme complexes (tenase complex and prothrombinase complex) catalyze thrombin generation.
- This is the thrombin burst phase, when enough Factor IIa is generated to induce plug formation.
- Thrombin generation subsequently converts fibrinogen into fibrin, which stabilizes the fibrin mesh at the site of vascular injury.2
- Clinical Pearls
- FXI: It is a critical enzyme for thrombin ignition. When plasma levels reach 20-40% of normal value it has a moderate effect on thrombin generation.
- Prothrombin (FII): The coagulation factor with the highest circulating concentration. It is an acute phase reactant which activates platelets, FXIII, and the antifibrinolytic pathway.
Hemostatic Plug
- Fibrin Formation
- Fibrinogen is converted into fibrin. FXIII is activated via FIIa and will cross-link the fibrin strands as they bind to their receptors on (GPIIb/IIIa) on the surface of activated platelets to initiate an expanding matrix that will become the hemostatic plug.
- Clinical Pearls
- FXIII: It is stored and released from platelets. In addition to cross-linking of the fibrin product, it catalyzes the binding of a2-antiplasmin to the strands, thereby conferring strength to the clot. Depletion of FXIII less than 50% is detrimental to clot sturdiness.2
Antifibrinolysis
- The function of the antifibrinolytic pathway is to confer resistance to the packed dense fibrin strands(Figure 3) .3
- Thrombin activatable fibrinolysis inhibitor (TAFI) is released by activated platelets, with the highest concentration near the site of the injury. It is rapidly activated by IIa and XIIIa and cross-linked to fibrin, providing further resistance to fibrinolysis.3
- Plasminogen activator inhibitor (PAI) is produced by platelets, endothelial cells, smooth muscle, and liver and is primarily stored in the platelets. It inhibits the catalytic site of tissue plasminogen activator (tPA).
- A2-antiplasmin is also released by platelets, and it prevents the plasminogen-mediated cleavage of fibrin.3
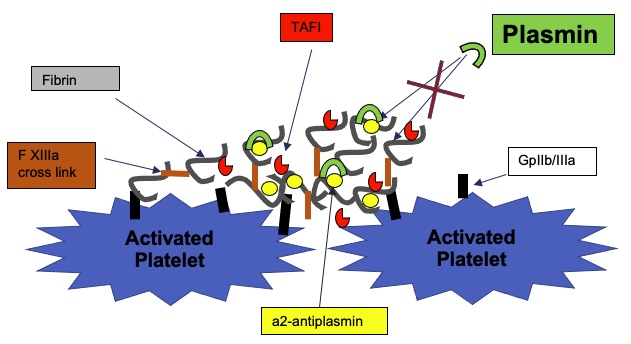
Figure 4. Antifibrinolytic pathway
Termination of Clotting
The rapid hemostatic response initiated by the clotting cascade must be regulated by multiple antithrombotic mechanisms to prevent thrombosis and tissue damage.
The main physiologic inhibitors of the coagulation pathway include:
- Tissue factor pathway inhibitor (TFPI): It is a serine protease inhibitor that is released by activated platelets and endothelial cells. It will bind TF:VIIa and FXa to prevent further progression of the coagulation cascade.
- Antithrombin III: It is a serine protease inhibitor that binds IIa and Xa in an equipotent fashion. Its activity is enhanced in the presence of endothelial heparan sulfate.
- Protein C (PC): It is a vitamin K-dependent protein. It is activated to APC when endothelial thrombomodulin binds IIa. APC forms a potent anticoagulant complex along with Protein S (PS) and deactivates Va. The formation of APC:PS:Va will dramatically increase the affinity of the structure for VIIIa, leading to its inactivation and removal from circulation.1,2,3
The termination phase of clotting is vital in mediating the extent of clot formation.
Patients with genetic deficiencies in the above pathways, such as Factor V Leiden mutation, deficiency in Protein C and S, and antithrombin III deficiency, are predisposed to clot formation given their hypercoagulable state.4
Fibrinolysis
- Fibrinolysis is a parallel system that is activated alongside the coagulation pathway to limit the size of clot formation via fibrin proteolysis. It is an enzymatic process that dissolves fibrin clots primarily through the conversion of plasminogen into its active proteolytic form plasmin3 (Figure 5).
- The two main classical enzymes that catalyze the conversion of plasminogen into plasmin are tissue-type and urokinase-type plasminogen activators (tPA and uPA, respectively). tPA is released from activated endothelium in response to endogenous chemokines/catecholamine release (epinephrine, vasopressin, and bradykinin), thereby making it an acute phase reactant. Once released, tPA binds to plasminogen on the fibrin surface or to PAI. It is precipitously removed from the circulation by the liver in the form of PAI-tPA complexes.
- Plasmin will seek lysine residues on the fibrin strands and cleave them, exposing further sites for plasminogen and plasmin to bind.
- D-dimers are produced by the digestion of cross-linked fibrin, which provides a clinical measure of fibrinolysis that may aid in the diagnosis of pulmonary embolism and DIC.
- D-dimer and a2-antiplasmin-plasmin complexes last for hours in circulation, potentially causing coagulation derangement.5
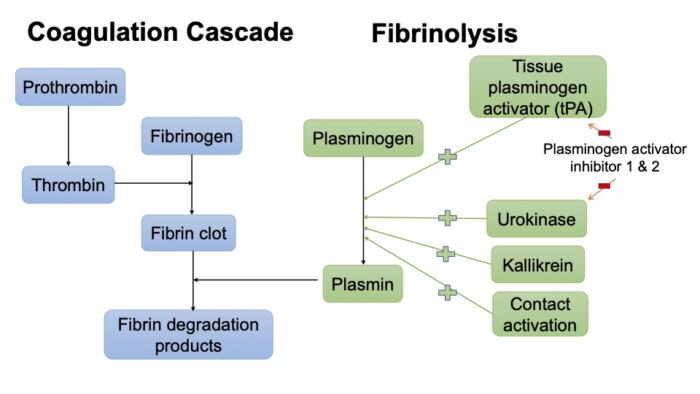
Figure 5. Simplified coagulation and fibrinolytic pathway
References
- O'Donnell J, O'Sullivan J, Roger JS. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br J Haematol. 2019;186(1):24-36. PubMed
- Bolliger D, Görlinger K, Tanaka KA. Pathophysiology and treatment of coagulopathy in massive hemorrhage and hemodilution. Anesthesiology. 2010; 113(5):1205-19. PubMed
- Moore EE, Moore HB, Kornblith LZ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers. 2021; 7(1):30. PubMed
- Bombeli T, Spahn DR. Updates in perioperative coagulation: Physiology and management of thromboembolism and haemorrhage. Br J Anaesth 2004; 93:275-87. PubMed
- Moore HB, Moore EE, Neal MD, et al. Fibrinolysis shutdown in trauma: Historical review and clinical implications. Anesth Analg. 2019; 129(3): 762–73. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.