Key Points
- ⍺-thalassemia and β-thalassemia are the two major forms of thalassemia, and both present clinically with microcytic, hypochromic anemia and, potentially, iron overload.
- The main treatments for the major forms of thalassemia are transfusion and iron chelation.
- Genetic counseling and screening are the primary prevention methods for thalassemia, and they can help treat the disease earlier in younger patients.
Introduction
- Thalassemias are a heterogeneous group of genetic disorders inherited in an autosomal recessive fashion. They are characterized by the absence or decreased synthesis of the alpha or beta globin chains of hemoglobin.
- Alpha-thalassemia is characterized by genetic deletions of one or both ⍺ globin chains. Nondeletions, caused by point mutations on the alpha-1 or alpha-2 globin gene, can rarely also be the cause.
- Βeta-thalassemia is one of the most common autosomal recessive disorders characterized by a lack of beta-globin chains. It can be caused by any of 200-plus point mutations, with deletions rarely being the cause.
- Cardiac failure is the primary cause of death in patients with any form of thalassemia due to the heart being overloaded with iron.
- Research is ongoing to find hepcidin up-regulators to avoid iron overload, as hepcidin can inhibit iron absorption.
Pathophysiology
- Alpha thalassemia is prevalent in tropical and subtropical world regions where malaria is still epidemic.
- Alpha-thalassemia affects patients through four clinical conditions: two carrier states and two major clinical forms of the disease, which include HbH disease and Hb Bart hydrops fetalis syndrome.1
- HbH disease is characterized by the presence of only a singular alpha globin gene, resulting in a surplus of beta-globin chains that comprise beta-4 tetramers; HbH is unstable inside red blood cells, leading to hemolysis in most patients.1
- Hb Bart hydrops fetalis syndrome is the gravest ⍺-thalassemia form, characterized by the absence of all four alpha globin genes. It affects the fetus by disrupting its ability to produce HbF or HbA.1 Clinically, severe anemia with Hb levels ranging from 3-8 g/dL is found with hepatosplenomegaly and heart failure.1
- Hb Bart hydrops fetalis can affect pregnancies, leading to preeclampsia, polyhydramnios, oligohydramnios, and death in-utero or shortly after birth. Currently, there is no effective treatment for Hb Bart hydrops fetalis.1
- Beta-thalassemia affects patients through three clinical conditions of increasing severity: a carrier state, thalassemia intermedia, and thalassemia major.
- Beta-thalassemia carrier state results from heterozygosity of beta-globin chains, and the patients are clinically asymptomatic.
- Thalassemia major is characterized by severe transfusion-dependent anemia.
- Thalassemia intermedia includes a heterogeneous group of disorders ranging in severity.

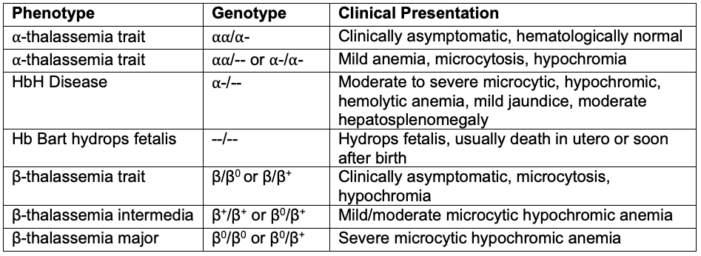
Table 1. Genotype and phenotype of variations of alpha and beta-thalassemia
- Beta-thalassemia, along with alpha-thalassemia, causes erythroid precursors to be five to six times more abundant in the bone marrow of patients, along with up to fifteen times the normal number of apoptotic cells.2 This leads to accelerated apoptosis, a major cause of defective erythropoiesis that stems from excess ⍺ chain deposition.3
- The primary clinical feature shared by ⍺ and β-thalassemia is iron overload.4 The excess of iron causes cardiac failure, the number one cause of death in thalassemia patients.4 Iron is connected to the heart’s rhythm, so it can cause abnormal contractions when it is too high or too low.
- Clinical studies exhibit patients with both forms of thalassemia having a mRNA inhibitor of hepcidin, which increases the absorption of iron in the small intestine.4 This would lead to the higher iron deposition that is seen in many patients’ organs of the gastrointestinal system, which results in organ failure in many cases.4
Clinical Presentation and Diagnosis
- The clinical presentation of thalassemia varies widely depending on the type and severity (Table 1).
- Alpha-thalassemia carriers are usually asymptomatic and don’t need any treatment.
- Patients with HbH disease present with moderate to severe microcytic, hypochromic anemia, mild jaundice, and moderate hepatosplenomegaly. A form of HbH disease associated with myelodysplasia has also been described.
- Carriers of beta-thalassemia are also asymptomatic.
- Infants with beta-thalassemia major present with severe anemia, failure to thrive, feeding problems, splenomegaly, etc., and require frequent blood transfusions.
- After about 10-11 years of age, these patients are at risk for complications related to iron overload. Complications commonly seen in adults with iron overload include:
- Heart: dilated cardiomyopathy, pericarditis
- Liver: chronic hepatitis, fibrosis, and cirrhosis
- Endocrine glands: diabetes, insufficiency of thyroid, parathyroids, pituitary, etc.
- Spleen: hypersplenism
- Bones: osteoporosis, skeletal changes
- Venous thrombosis
- The diagnosis of thalassemia is suspected in patients with microcytic and hypochromic hemolytic anemia on blood smears, jaundice, hepatosplenomegaly, and bone alterations (Table 2). Anisocytosis, poikilocytosis, and nucleated red blood cells are additional findings on blood smears.5

Table 2. Lab values associated with ⍺ and β-thalassemia
- Thalassemia major is diagnosed by more severe symptoms than those seen in all forms. Thalassemia minor is diagnosed as the carrier state because most patients are asymptomatic.5 Thalassemia intermedia has findings that range between the two extremes of the carrier state and the major state.5
Treatment
- Patients with thalassemia major usually receive regular blood transfusions (every 2-3 weeks) to correct the anemia. A pretransfusion hemoglobin of 9.5-10 g/dL is commonly used as a transfusion threshold.
- To reduce the risk of iron overload, chelation therapy is often initiated after 10-12 transfusions with chelating agents such as deferoxamine, deferasirox, and deferiprone.
- Hematopoietic stem cell transplantation is a potential option in selected cases.8
- Allogenic stem cell transplantation (alloHSCT) has been shown to significantly impact survival in thalassemia patients.9 When alloHSCT is utilized early in the disease process before iron overload and transfusion complications occur, up to 90% survival rates have been reported. Survival rates without HSCT have not been distinctly reported, but they would be lower without the treatment in general as end-organ damage would continue.
- Gene therapy is the latest advancement in severe thalassemia management.
- A new attractive clinical application is using designer nucleases to induce HbF.9 This approach can disrupt the target sequences in hematopoietic stem/progenitor cells through nonhomologous end joining. This would present a new cure for thalassemic patients, as the ability to edit the gene and repress/activate globin repressors would now be an ideal clinical approach.
- Patients with thalassemia may undergo a splenectomy to limit the number of transfusions or to treat hypersplenism.
Perioperative Management
- Perioperative care of patients with thalassemia is challenging due to iron overload, anemia, and multiple organ involvement.10
- Patients with thalassemia may present for elective or urgent surgeries, including therapeutic splenectomy, cholecystectomy for gall stones, correction of facial deformities and debridement of leg ulcers, fixation of fractures, etc.10
- A comprehensive preoperative evaluation and multidisciplinary approach are critical.
- There are no specific guidelines for ideal perioperative hemoglobin levels. The Royal College of Obstetricians and Gynaecologists considers ten g/dL a safe and appropriate level.6 Patients with low hemoglobin levels should undergo blood transfusion preoperatively due to blood loss.6
- There is no recommendation for discontinuing iron chelation therapy before surgery.10
- In thalassemic patients with facial defects, the airway should be carefully evaluated, as maxillary hypertrophy can interfere with intubation at a rate of up to 19%.6 Also, high-arched palates present a problematic issue with placing supraglottic airways.6,10
- In addition to standard American Society of Anesthesiologists monitors, advanced hemodynamic monitoring such as transthoracic echocardiogram and invasive arterial blood pressure monitoring should be considered based on the patient’s comorbidities.6,10
- The choice of anesthetic technique and drugs depends on the surgery and patient comorbidities. Both general and regional anesthesia have been successfully used. Neuraxial techniques may be challenging secondary to spinal skeletal abnormalities such as scoliosis and osteoporosis.10 Preoperative coagulation tests should be performed before a neuraxial block.
- Hypercoagulability is common, and appropriate deep vein thrombosis (DVT) prophylaxis should be used. These patients are frequently immunocompromised and prone to infections. Therefore, perioperative antibiotic prophylaxis should be used.10
- The perioperative management of patients with thalassemia is summarized in Table 3.
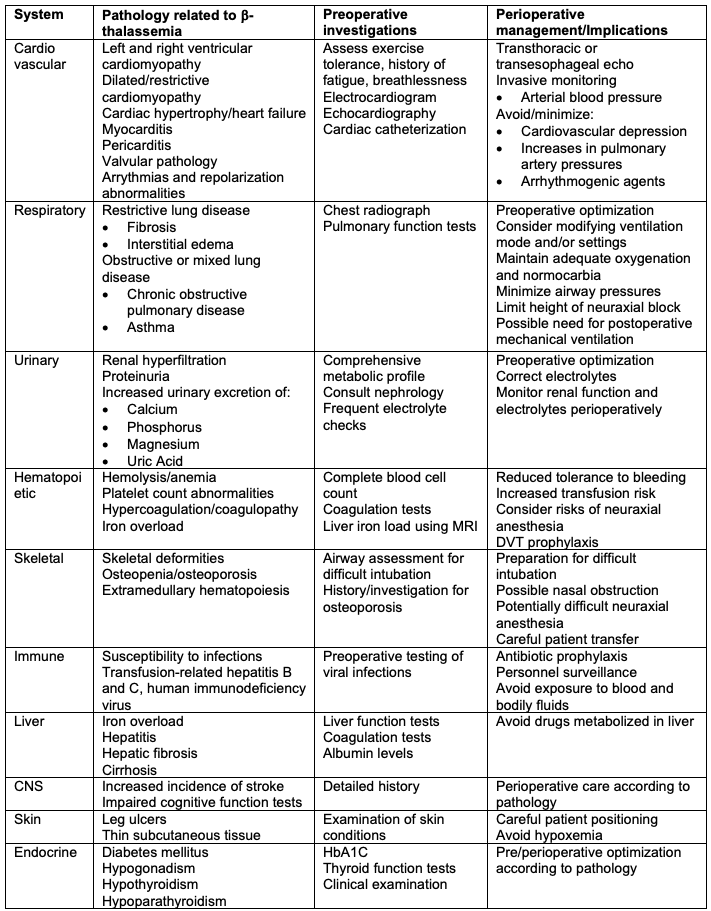
Table 3. Perioperative implications and management of β-thalassemia patients. Adapted from Staikou C et al. Anaesthesia. 2014.10
References
- Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med. 2011;13(2):83-88. PubMed
- Centis F, Tabellini L, Lucarelli G, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96(10):3624-9. PubMed
- Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood. 2000;96(9):2606-12. PubMed
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135-46. PubMed
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76. PubMed
- Singh A, Sharma K, Venkateswaran V, et al. Pregnancy in thalassemia. Anesthetic implications and perioperative management. A narrative review. Journal of Obstetric Anaesthesia and Critical Care. 2021;11(2):81-9. Link
- Wood JC. Cardiac complications in thalassemia throughout the lifespan: Victories and challenges. Ann NY Acad Sci. 2023;1530(1):64-73. PubMed
- Srivastava A, Shaji RV. Cure for thalassemia major - from allogeneic hematopoietic stem cell transplantation to gene therapy. Haematologica. 2017;102(2):214-223. PubMed
- Azhagiri MKK, Babu P, Venkatesan V, Thangavel S. Homology-directed gene-editing approaches for hematopoietic stem and progenitor cell gene therapy. Stem Cell Research & Therapy, 2021;12(1):500. PubMed
- Staikou C, Stavroulakis E, Karmaniolou I. A narrative review of peri-operative management of patients with thalassaemia. Anaesthesia. 2014;69(5): 494-510. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.