Key Points
- Hemophilia A and B are congenital coagulation disorders caused by a deficiency or absence of either factor VIII (FVIII) or factor IX (FIX).
- With multidisciplinary planning, surgery can be safe and successful for patients with hemophilia.
- Factor concentrates, plasma-derived or recombinant, are the mainstay of hemophilia treatment. Thromboprophylaxis should be considered on an individual basis, especially if the factor is being replaced to almost normal levels.
- Adequate hemostasis is critical to proper healing and avoidance of complications such as hematoma formation, infection, and wound dehiscence.
Introduction
- Hemophilia A and B are congenital coagulation disorders caused by a deficiency or absence of either of two coagulation proteins, FVIII or FIX.1
- Hemophilia A is caused by a deficiency in FVIII, and hemophilia B is caused by a deficiency in FIX.
- Currently, only hemophilia A and B are defined as hemophilias.1
- Both hemophilia A and B are inherited as X-linked recessive disorders. Thus, the majority of people affected by hemophilia are male.1
- Women are usually heterozygous carriers of a mutated gene, usually have decreased FVIII or FIX levels, and can present with mild symptoms.1
- However, women and girls who are hemophilia carriers are at risk for bleeding symptoms, including heaving menstrual periods and musculoskeletal bleeding.2
- Hemophilia C or Rosenthal disease is caused by a deficiency in factor XI.
- It is inherited in an autosomal pattern and presents with variable bleeding tendency despite severely depleted FXI levels.3
- Severe disease is characterized by less than 20% of normal FXI levels.
- Acquired Hemophilia A is a rare autoimmune bleeding disorder characterized by the development of neutralizing autoantibodies against FVIII.4
- It can manifest in both men and women with no prior bleeding history.
- Its incidence increases significantly with age.4
Pathophysiology1-3
- Due to the deficiency or absence of specific coagulation factors, the body is unable to initiate and amplify the clotting cascade, leading to a weak fibrin clot and prolonged bleeding.
- Deficiency of either FVIII or FIX results in compromised factor X activation, resulting in suboptimal thrombin generation and inadequate early clot strength.1
Genetics and Epidemiology
- The genetics and epidemiology of hemophilia A, B, and C are listed in Table 1.

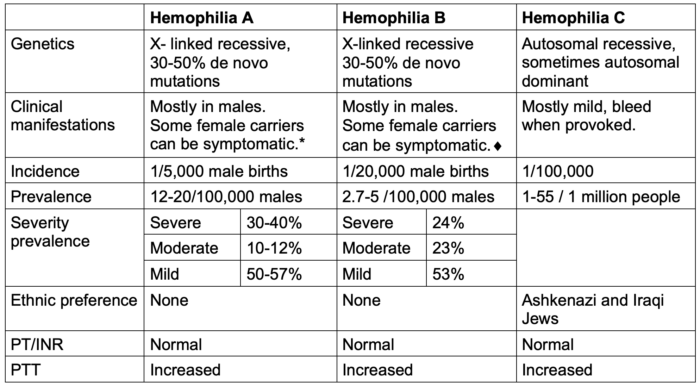
Table 1. Genetics and epidemiology of hemophilia A, B, and C.
*Severe bleeding in females can occur when 1 X chromosome is missing, e.g., in Turner’s syndrome (XO).
♦︎Symptomatic carrier is defined as an International Society on Thrombosis and Hemostasis- Bleeding Assessment Tool score ≥ 6 for teenage and adult females and ≥3 for young girls.5 The severity of bleeding in a carrier does not always correlate with residual factor activity.
Abbreviations: PT, prothrombin time; INR, international normalized ratio; PTT, partial thromboplastin time
Clinical Presentation
- Hemophilia A and B have similar symptoms and are characterized by bleeding (spontaneous or after trauma) into large joints, such as elbows, knees, and ankles. This causes painful hemophilic arthropathy.1
- Patients with severe hemophilia may have serious, life-threatening bleeds, such as intracranial bleeds and bleeds in other internal organs.
- Patients with hemophilia C have variable bleeding tendencies despite severely deficient FXI levels, which is most commonly found in homozygotes.3
- The clinical presentation of hemophilia is summarized in Figure 1.
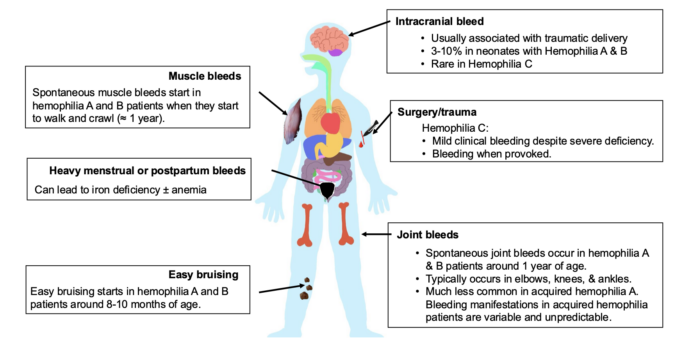
Figure 1. Clinical presentation of hemophilia
Diagnosis
- The diagnosis of hemophilia is based on three main clinical scenarios.1
- Positive family history
- Abnormal bleeding tendencies
- Unexpected abnormal coagulation tests
- Typically, children with moderate or severe hemophilia show easy bruising around 8-10 months of age, especially when they start crawling and/or walking.
- If untreated, patients with severe disease have an average of 25-50 bleeding events per year, while patients with moderate disease have an average of 5-10 events per year, and patients with mild disease have 1-2 bleeding events per year.
- The laboratory diagnosis is based on measuring FVIII and FIX clotting activity.
- The severity of disease for hemophilia A and B is based on factor levels and residual FVIII and FIX activity:
- Mild = 6 – 40 IU/dL
- Moderate = 1 – 5 IU/dL or 1-5% of factor activity
- Severe <1 IU/dl or <1% of factor activity
- Additionally, activated partial thromboplastin time (aPTT) is prolonged, and prothrombin time (PT) and international normalized ratio (INR) are normal.
- Genetic analysis is often performed to identify the causative mutation.
Treatment6,7
Factor Concentrates
- Factor concentrates are the mainstay of treatment for hemophilia A and B. There are three broad approaches to factor replacement therapy depending on hemophilia severity.1
- On-demand therapy: replacement factor given at the time of bleeding, including breakthrough bleeding while on continuous prophylaxis.
- Intermittent prophylaxis: replacement factor given to prevent bleeding for short periods of time, such as during and after surgery.
- Continuous prophylaxis: replacement factor given to prevent bleeding for at least 45 of 52 weeks of a year.
- Primary prophylaxis: continuous prophylaxis started before 3 years of age and before the first or second large joint bled
- Secondary prophylaxis: continuous prophylaxis started after two or more large joint bleeds.
- Tertiary prophylaxis: continuous prophylaxis started at the onset of arthropathy.
- Primary prophylaxis is the current goal but is limited by resources in some countries. Frequency of prophylactic dosing depends on the factor product and the frequency needed to minimize breakthrough bleeding.
- The World Federation of Hemophilia (WFH) recommends factor dosing goals based on the type of bleeding or surgery.6 Table 1 below provides an example of factor dosing goals for patients with hemophilia A. Several factor concentrates, either plasma-derived or recombinant factors, are available for hemophilia A and B.
- Recombinant factor concentrates are also available with a standard half-life (10-14 hours for FVIII and 22-26 hours for FIX) or an extended half-life (14-48 hours for FVIII and 86-118 hours for FIX).
- Of note, the half-lives of FVIII and FIX are age-dependent, with half-lives being longer in adults than in children.
- The required dose for FVIII concentrates (IU) = body weight (kg) x desired rise in FVIII activity (% activity) x 0.5.
- A general rule of thumb for FVIII dosing is that for every IU of FVIII administered per kg of body weight, FVIII activity increases by 2%.7
- The required dose for FIX concentrates (IU) = body weight (kg) x desired increase in FIX activity (% activity).
- A general rule of thumb for FIX dosing is that for every IU of FIX administered per kg of body weight, FIX activity increases by 1%.7
- The hourly infusion rate, if needed, is calculated using the following formula.7
Infusion rate (IU/kg/hour) = clearance (mL/kg/hr) x target level (IU/mL) - There are currently no FDA-approved FXI products in the United States.
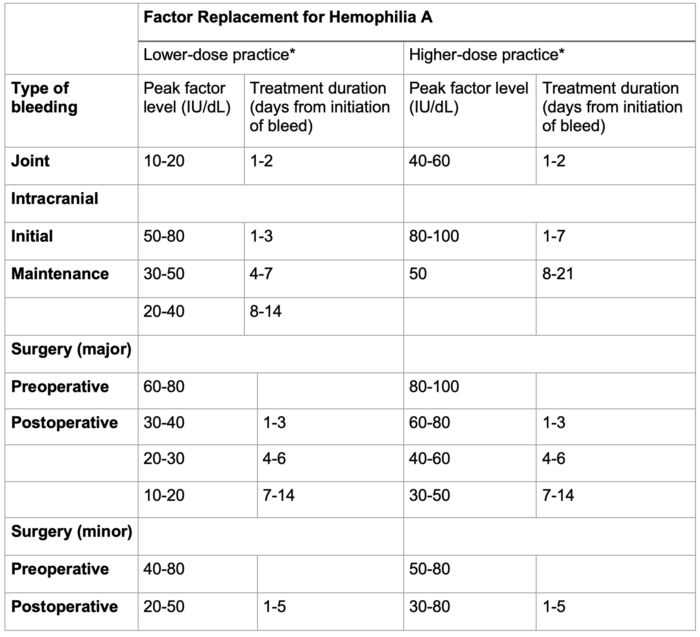
Table 2. Factor replacement for hemophilia A. Adapted from the World Federation of Hemophilia guidelines.6 Joint bleeds need much lower and shorter factor replacement than intracranial bleeding and major surgery. *Lower-dose and higher-dose regimens are included to represent global practice patterns influenced by resource availability.
Factor Concentrate Alternatives
- The use of cryoprecipitate (for hemophilia A and B) and fresh frozen plasma (for hemophilia C) is limited by the large volumes required due to the relatively low concentration of factors in these blood products.
- Factor eight inhibitor bypassing agent (FEIBA): FEIBA is an activated 4-factor prothrombin complex concentrate containing factors II, VIIa, IX, and X that is used as a bypassing agent to treat patients with hemophilia A and B with inhibitors as well as acquired hemophilia A.
- Recombinant activated factor VII (FVIIa): Recombinant FVIIa was created as a bypassing agent to treat patients with hemophilia A and B with inhibitors. It can also be used to treat patients with acquired hemophilia A.
- Emicizumab: Emicizumab is a monoclonal antibody designed to mimic the function of FVIII (facilitates activation of FX by FIX). It was initially created to treat patients with hemophilia A with inhibitors, but its use has since expanded to prophylaxis for patients with hemophilia A without inhibitors. It is not useful for treating active bleeding. Its popularity is based on its subcutaneous route and long half-life (30 days), which allows for infrequent dosing (weekly to monthly). Its main drawback is its interference with tests that include aPTT, FVIII level, FVIII inhibitor titer, and activated clotting time.
- Gene therapy: Multiple products have been investigated, with the goal of introducing a viral vector carrying the gene for FVIII or FIX in a single dose. Hemophilia A and B each have an FDA-approved product. Both long-term safety and durability are still being monitored.
Complications
- Inhibitory antibodies (inhibitors):
- A complication of replacement therapy is IgG antibodies against FVIII (hemophilia A) or FIX (hemophilia B), which make endogenous and exogenous FVIII or FIX ineffective. Bypassing agents such as FEIBA and recombinant-activated Factor VII are needed to prevent and/or treat bleeding.
- Patients with hemophilia A (30%) are more prone to inhibitors than those with hemophilia B (5-10%).
- About 10% of patients with severe hemophilia C develop antibodies.
- Chronic pain:
- Mostly in indexed/target joints (knees, elbows, and ankles)
- Recurrent bleeding leads to severe joint destruction.
- Poor quality of life due to painful arthropathy, reduced mental wellness, and stress have improved remarkably due to the availability of factor products and the practice of prophylactic replacement therapy.2
Perioperative Considerations
- Multidisciplinary collaboration, including surgery, anesthesiology, hematology, blood bank, laboratory, pharmacy, and physical therapy, is needed for individualized planning throughout the perioperative period.
- A hematologist’s treatment plan includes intraoperative and postoperative factor goals, adjuncts such as antifibrinolytics and desmopressin, and monitoring strategies.
- Factor assays: One-stage clotting assays for FVIII and FIX are commonly used and have a turn-around time of 1-2 hrs.
- There are no formal recommendations for the use of viscoelastic testing.
- Multimodal pain management: Neuraxial and regional anesthesia are possible with factor replacement to >50 IU/dL and normal coagulation tests.
- Consider thromboprophylaxis on an individual basis; it is likely needed if maintaining close to normal factor levels.
- Good hemostasis for 2-3 weeks postoperatively is necessary for proper healing and avoidance of complications such as hematoma formation, infection, and wound dehiscence.
References
- Berntorp E, Fischer K, Hart DP, et al. Haemophilia. Nat Rev Dis Primers. 2021;7(1):45. PubMed
- Weyand AC, Sidonio RF, Jr., Sholzberg M. Health issues in women and girls affected by haemophilia with a focus on nomenclature, heavy menstrual bleeding, and musculoskeletal issues. Haemophilia. 2022;28 Suppl 4(Suppl 4):18-25. PubMed
- Lewandowska MD, Connors JM. Factor XI Deficiency. Hematol Oncol Clin North Am. 2021;35(6):1157-1169. PubMed
- Lehoczki A, Fekete M, Mikala G, et al. Acquired hemophilia A as a disease of the elderly: A comprehensive review of epidemiology, pathogenesis, and novel therapy. Geroscience. 2024; 47(1):503-14. PubMed
- Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20(6):831-5. PubMed
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1-158. PubMed
- Kwak J, Mazzeffi M, Boggio LN, Simpson ML, Tanaka KA. Hemophilia: A review of perioperative management for cardiac surgery. J Cardiothorac Vasc Anesth. 2022;36(1):246-25. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.