Key Points
- Duchenne muscular dystrophy (DMD) is a lethal neuromuscular disorder characterized by progressive, symmetrical proximal muscle weakness that typically manifests in males and is caused by an X-linked recessive mutation on the X chromosome, which prevents the normal formation of dystrophin, a muscle-stabilizing protein.1
- The complete loss of dystrophin in DMD and partially functional dystrophin in Becker muscular dystrophy (BD) disrupts sarcolemmal integrity and leads to myofibril atrophy, necrosis, and fibrosis.
- Although patients with DMD and BD are not at an increased risk for malignant hyperthermia (MH) compared to the general population, they are prone to rhabdomyolysis, life-threatening hyperkalemia, and cardiac arrest after administration of succinylcholine or with exposure to volatile anesthetics.2
Introduction
- DMD is a lethal neuromuscular disorder characterized by progressive, symmetrical proximal muscle weakness that typically manifests in males and is caused by an X-linked recessive mutation on the X chromosome, which prevents the normal formation of dystrophin, a protein that stabilizes muscle.1
- DMD is a common genetic disorder with an incidence of 1 in 3500 male births, with muscle weakness onset usually by age 5 years and progression to wheelchair dependence by age 14 years. An affected child might present with delayed walking, difficulty running or jumping, pseudohypertrophy of the calves, pronounced lumbar lordosis, and elevated creatine phosphokinase levels (Figure 1, Table 1). The classic Gowers sign is characterized by the use of the arms when rising from the floor.3 Proximal muscles are affected earlier than distal limb muscles. Usually, the lower extremity muscles are affected before the upper extremity muscles.4

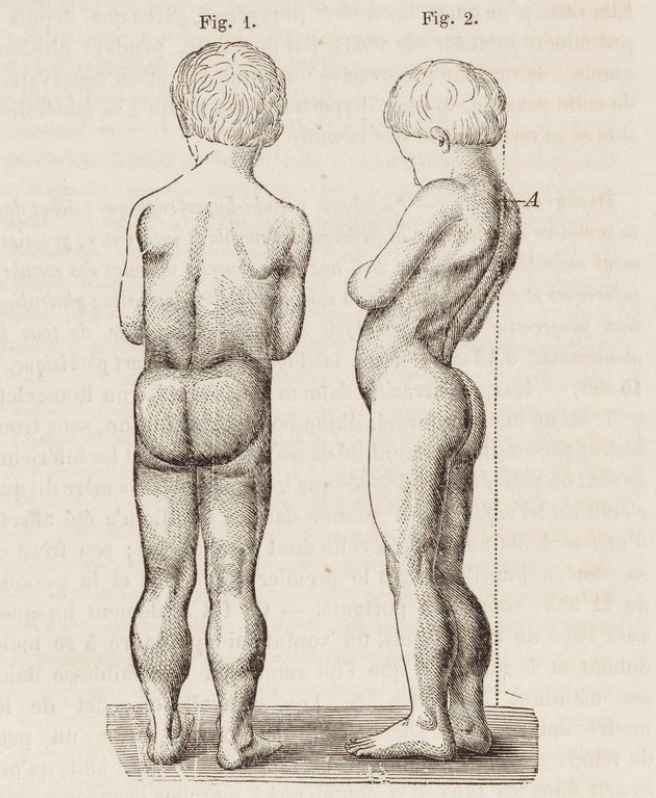
Figure 1. Drawing of the typical features of a boy with Duchenne muscular dystrophy. Note the pseudohypertrophy of the calves, thinness of the arms, and lumbar hyperlordosis. Source: Duchenne, Guillaume-Benjamin - 1868. Arch. Gen. Med. Bibliothèque Nationale de France. Public Domain via Wikimedia Commons. Link
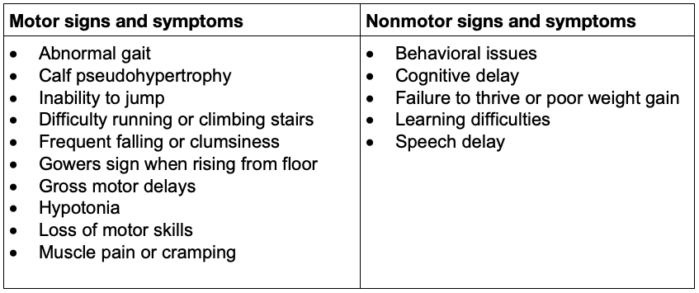
Table 1. Common early signs and symptoms in patients with DMD3
- Until recently, most patients with DMD did not survive much beyond their teen years. With advances in cardiac and respiratory care, life expectancy is increasing into the 30s. Most common causes of death include progressive cardiomyopathy, ventilatory pump insufficiency, and pneumonia.1,4
- Becker dystrophy affects one in 30,000 male births, with proximal leg and pelvis muscle weakness onset usually in teens or early adulthood, and slower, less predictable progression. Dilated cardiomyopathy, not consistent with the patient’s skeletal muscle weakness, may be the initial presentation. Mortality in BD usually occurs between the ages of 30 and 60 and is caused by respiratory or heart failure.2
- Both DMD and BD are caused by an X-linked recessive mutation (Figure 2) in the dystrophin gene, which encodes the protein responsible for stabilizing the sarcolemma. The dystrophin gene is the largest known human gene, containing 79 exons. Two-thirds of all DMD mutations are inherited from the mother, who is a carrier. At the same time, one-third are de novo mutations, with whole-exon deletions or duplications being the most common (70%).3
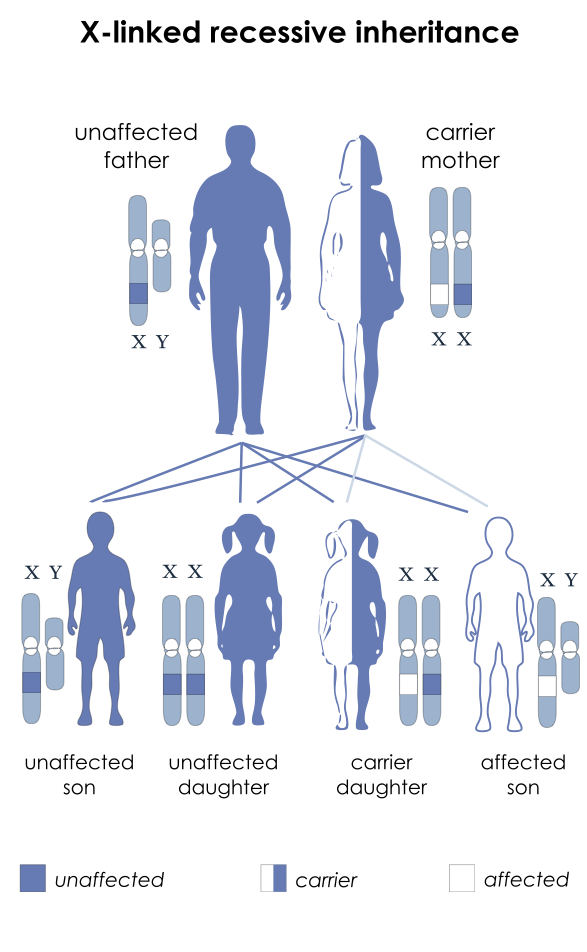
Figure 2. Inheritance pattern of X-linked recessive diseases such as DMD and BD. The mutation is passed to the male offspring from the mother who is a carrier. Source: National Institutes of Health, Public Domain, via Wikimedia Commons. Link
- Dystrophin helps anchor the actin of the contractile apparatus inside the muscle cell to the layer of connective tissue surrounding each muscle fiber. It acts as a shock absorber during contraction, preventing muscle damage (Figure 3). Dystrophin is also important in organizing postsynaptic nicotinic acetylcholine receptors (nAChRs). In the absence of normal muscle dystrophin, abnormalities occur in the number, type, and location of these receptors, which may contribute to the instability of the muscle membrane. Mutations in the central nervous system dystrophin isoform are associated with behavioral issues, cognitive delay, and learning difficulties (Table 1). Dilated cardiomyopathy is an effect of cardiac isoform abnormalities.
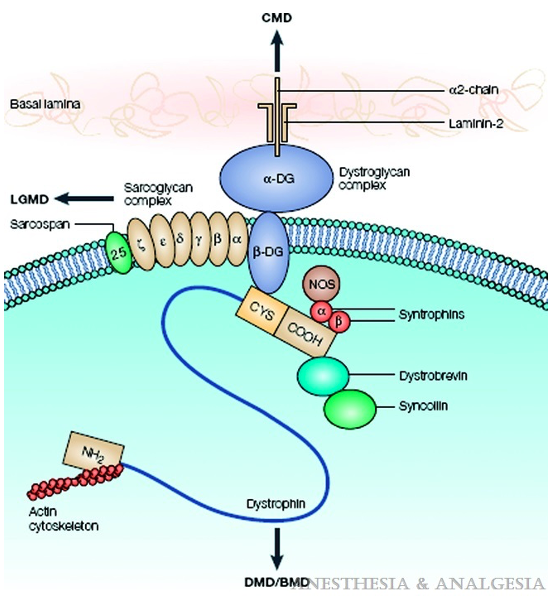
Figure 3. Dystrophin, as part of the dystrophin-glycoprotein complex, links the actin of the contractile apparatus inside the muscle cell to the connective tissue laminin in the extracellular matrix, maintaining sarcolemmal stability. Arrows indicate dysfunction of which elements of the complex lead to which type of muscle dystrophy. Abbreviations: DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; CMD, congenital muscular dystrophy; CYS, cysteine; DG, dystroglycan; LGMD, limb-gridle muscular dystrophy; NOS, nitric oxide synthase. Source: Malignant hyperthermia and muscular dystrophies Anesth Analg. 2009;109(4):1043-8.
- Complete absence of dystrophin in DMD and a qualitative and quantitative dysfunction in BD lead to a repeat cycle of muscle injury, necrosis, regeneration, and replacement of dead muscle cells by fibrosis and fat infiltrates (Figure 4).
- As a result of muscle regeneration, postsynaptic nAChRs are expressed as a mixture of fetal and mature receptors. Because of the presence of fetal receptors, administration of succinylcholine is contraindicated as it may trigger rhabdomyolysis and potentially fatal hyperkalemia.2
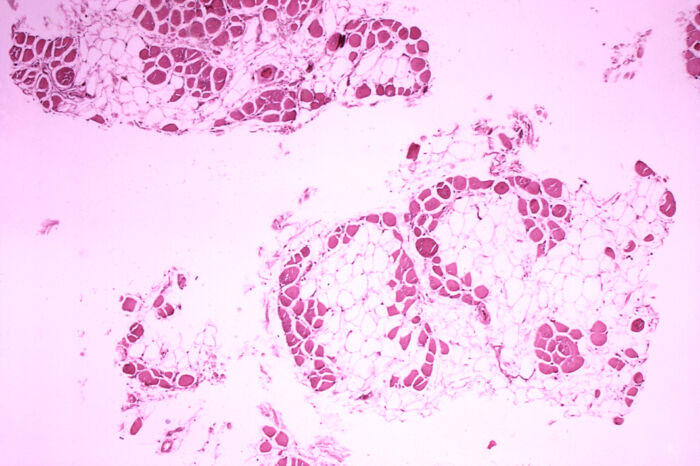
Figure 4. Microscopic cross-section of the gastrocnemius muscle in a patient with DMD showing extensive replacement of muscle fibers with fat cells. Source: Centers for Disease Control and Prevention’s Public Health Image Library, Public Domain via Wikipedia Commons. Link
Diagnosis
- Muscle weakness, not walking by 16-18 months of age, Gowers sign, toe-walking (any age, especially less than 5 years of age), and clumsiness in young boys should prompt an evaluation of serum creatine kinase (CK) levels, which is elevated in DMD due to leakage into the bloodstream from damaged muscle cells.3
- An elevated CK level should lead to a neuromuscular consultation and DNA testing for mutations in the DMD gene, using either multiplex ligation-dependent probe amplification analysis or comparative genomic hybridization array.
- If genetic testing does not confirm the clinical diagnosis of DMD, protein analysis on muscle biopsy using immunohistochemistry or Western blotting is done to evaluate the size, location, and amount of dystrophin.3
- CK level does not correlate with the severity of the disease and tends to decrease with the loss of muscle mass.
Respiratory Comorbidities
- DMD is characterized by progressive weakness of the diaphragm, intercostal muscles, and the accessory muscles of respiration, resulting in restrictive lung disease (Figure 5) and progressive decrease in total lung capacity and vital capacity. Expiratory muscles are affected first.1,5
- Scoliosis further impairs pulmonary function.
- Hypoventilation and impaired cough lead to atelectasis and pneumonia.
- Respiratory failure commonly occurs in the 20s.
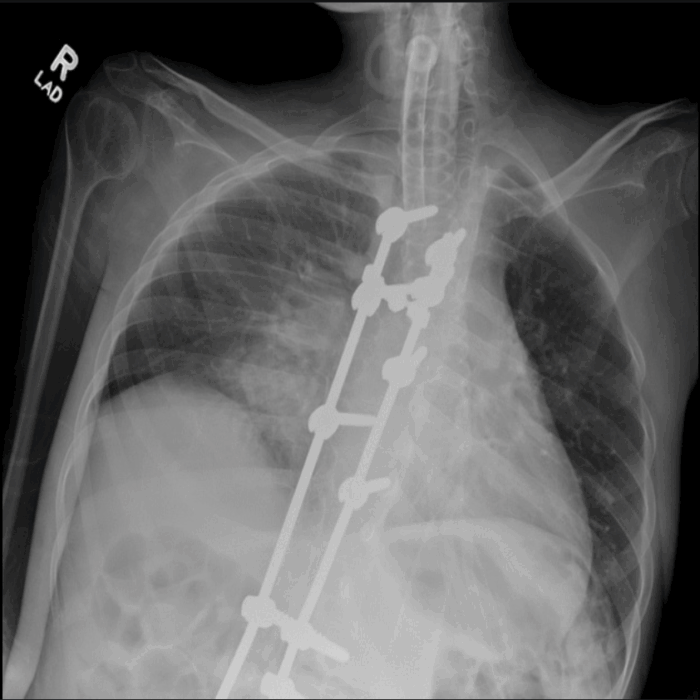
Figure 5. Chest X-ray of a 20-year-old tracheostomy-dependent male with DMD. Note low lung volumes, poor inspiratory effort, and airspace opacities in the right perihilar region, which may represent atelectasis, pneumonia, or aspiration. Posterior spinal fusion rods are seen in place. The visualized humeral shaft is thin and gracile with diffuse muscular atrophy. Source: Case courtesy of Kathryn Kinser, Radiopaedia.org, rID 66153. Link
Cardiac Comorbidities
- Myocardial degeneration and gradual replacement with fibrosis result in conduction abnormalities, valvular heart disease, and progressive dilated cardiomyopathy, which is present in one-third of DMD patients by age 14 years and almost all patients by age 18 years.2,6
- The American Academy of Pediatrics recommends that children with DMD undergo an initial cardiac evaluation, including an electrocardiogram (ECG) and echocardiogram, in early childhood. This evaluation should be repeated every other year until age 10, followed by annual evaluations thereafter.6
- ECG findings in DMD and BD include sinus tachycardia related to autonomic dysfunction, decreased rate variability, tall R waves in the right precordial leads, prominent left precordial Q waves, inverted T waves from left ventricle scarring, atrioventricular blocks, and bundle branch blocks.
- Typical echocardiographic findings in dilated cardiomyopathy associated with DMD include mitral valve prolapse, posterobasal hypokinesia in a thin-walled ventricle, and a slow relaxation phase with normal contraction.
- Patients with DMD and BD have limited ability to increase cardiac output in response to stress and are at increased risk of inadequate oxygen delivery during and after surgery.
Medical Management
- The care of patients with DMD includes a multidisciplinary approach involving pulmonary, cardiac, psychosocial, surgical management, and physical therapy.3
- Corticosteroids remain the mainstay of therapy for increasing muscle strength, preserving ejection fraction, and delaying the need for noninvasive ventilation. Side effects include weight gain, growth retardation, bone demineralization, hypertension, adrenal insufficiency and increased fracture risk.3,7,8
- Treatment with angiotensin-converting enzyme inhibitors and beta blockers can slow cardiac muscle degeneration if initiated as soon as echocardiography abnormalities are identified.
- Gene therapy targeting specific DMD variants is gaining popularity.8
- Givinostat is a deacetylase inhibitor that counteracts the downstream effects of dystrophin deficiency, reducing inflammation and fibrosis, and is FDA-approved for DMD patients who are 6 years or older.9
Anesthetic Considerations
- Patients with DMD and BD may require anesthesia for muscle biopsy, bronchoscopy, tracheostomy, gastric tube placement, correction of scoliosis, and release of contractures.
- Obesity is common in older boys due to immobility and steroid use. Hypertrophy of the tongue may cause airway obstruction. Limb contractures can make vascular access and positioning difficult, and spinal deformity may affect respiratory function and airway anatomy.10
- Anesthesia risk is increased in patients with DMD and involves potentially fatal reactions to succinylcholine and volatile anesthetics, upper airway obstruction, hypoventilation, atelectasis, intraoperative congestive heart failure, arrhythmias, respiratory failure, and difficulty separating from the ventilator.2
- Delayed gastric emptying increases the risk of pulmonary aspiration and requires expedient control of the airway.
- Perioperative complications may occur even in mildly affected patients.
Preoperative Evaluation1,2,5,6,10
- Meticulous preoperative evaluation and anesthesia planning are essential.
- Patients with DMD and BD should undergo pulmonary and cardiac evaluation before every anesthetic. Medical therapy of cardiac dysfunction should be optimized.
- Preoperative respiratory function should be assessed clinically and using pulmonary function measurements, including forced vital capacity (FVC), maximum inspiratory pressure, maximum expiratory pressure, and peak cough flow. Arterial blood analysis is required when the patient is not able to blow into the pulmonary function measuring device. A higher-than-normal arterial carbon dioxide tension is a late finding that indicates a poor prognosis.
- Preoperative cardiac evaluation should include an ECG and echocardiography. If the echocardiography shows abnormal findings, a dobutamine stress test should be considered.
- Preoperative laboratory values should include baseline measurements of potassium, calcium, magnesium, and creatinine phosphokinase levels. CK levels do not correlate with the severity of the disease because they tend to decrease with the loss of muscle mass.
Intraoperative Considerations1,2,10
- Inhaled anesthetics may cause rhabdomyolysis, intraoperative hyperthermia, tachycardia, and hyperkalemia-related cardiac arrest and should be avoided.
- Total intravenous anesthesia with propofol, ketamine, and a short-acting narcotic like remifentanil is preferred.
- Succinylcholine is contraindicated as it may cause rhabdomyolysis and life-threatening hyperkalemia. The FDA issued a warning against the use of succinylcholine in the pediatric population because of the potential for mortality in children with undiagnosed myopathies.
- Nondepolarizing muscle relaxants have a delayed onset and prolonged action. It’s recommended to use short-acting agents and monitor neuromuscular blockade with quantitative monitoring. Cholinesterase inhibitors are safe, and sugammadex has been used successfully.
- Medications that depress cardiac contractility or increase the likelihood of arrhythmias should be avoided, as contractility and conduction are already abnormal in these patients.
- Regional anesthesia techniques and multimodal analgesia should be used whenever possible.
- Opioids should be used cautiously as they may worsen the respiratory depression.
- Gurnaney et al. reviewed case reports of rhabdomyolysis and hyperkalemia-related intraoperative cardiac arrest when inhaled anesthetics were used and discovered that tachycardia/bradycardia typically preceded complete cardiovascular collapse.2 The risk of cardiac arrest was not related to the duration of inhaled anesthetic exposure. Marked hyperkalemia and metabolic acidosis were consistently identified during resuscitation.
- Patients with DMD undergoing scoliosis surgery have higher reported blood loss and a higher risk of intraoperative heart failure. This blood loss may be related to the inadequate vasoconstrictive response in the absence of dystrophin, the reduction in neuronal nitric oxide synthase, and platelet dysfunction.
- Use of intraoperative invasive blood pressure monitoring is recommended for surgeries associated with the risk of significant blood loss.
- Extubation directly to non-invasive positive pressure ventilation (NPPV) is recommended for DMD patients with a baseline FVC of less than 50% predicted, and is essential for those with an FVC of less than 30% predicted, as well as for patients who use NPPV preoperatively.5
DMD, BD, and the Risk of MH2
- Patients with DMD or BD are not at an increased risk of MH compared with the general population.2
- It is unlikely that a true genetic association exists between DMD and MH. The genetic mutation in DMD is located on the X chromosome, and MH-associated mutations are usually found on chromosome 19.2
- Literature reports exist on DMD patients testing positive on caffeine-halothane contracture test, but the validity of contracture testing in patients with muscular dystrophy has been debatable.
- Acute rhabdomyolysis and hyperkalemia reported in DMD patients are not typically associated with a hypermetabolic state.
- Succinylcholine administration should be avoided because of the risk of rhabdomyolysis, life-threatening hyperkalemia, cardiac arrest, and unsuccessful resuscitation.2
- Postulated mechanisms for succinylcholine-induced hyperkalemia include excess potassium release from up-regulation of abnormal extrajunctional acetylcholine receptors and development of hyperkalemia.2
- Exposure to volatile anesthetics in patients with DMD or BD may be associated with life-threatening rhabdomyolysis or cardiac complications. Therefore, inhaled anesthetics should be used cautiously or when their benefits outweigh the risks.2
- The exact pathophysiology of inhaled anesthetic-induced rhabdomyolysis is not known. A proposed mechanism is that inhaled anesthetics exacerbate the breakdown of already frail and vulnerable muscle membranes from movement and reversal drugs.2
Postoperative Considerations6
- DMD and BD patients should recover from surgery in the intensive care unit.
- Significant postoperative respiratory insufficiency is common and may occur up to 36 hours after surgery.
- Untreated pain may worsen already compromised pulmonary mechanics. Opioids should be used with caution.
- Rhabdomyolysis, hyperkalemia, and cardiac arrest may suddenly occur in the postoperative period after an uneventful intraoperative course and may be fatal.
- Postoperative anemia and inadequate volume replacement may compromise ventricular preload and oxygen delivery. Maintenance of fluid balance and cardiopulmonary monitoring are critical.
- Prothrombotic effects of muscle degeneration are associated with an increased risk of thrombotic events. Anticoagulation should be considered.
References
- Castro De Oliveira BM, Vazques R. Anesthesia for Orthopedic Surgery. In: Gropper MA, Miller RD. Miller's Anesthesia. 9th ed. Philadelphia, PA: Elsevier; 2020: 1952-83.
- Gurnaney H, Brown A, Litman RS. Malignant hyperthermia and muscular dystrophies. Anesth Analg. 2009;109(4):1043-8. PubMed
- Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-67. PubMed
- Muscular Dystrophy Association. Duchenne Muscular Dystrophy (DMD) Disease. Accessed date: January 2025. Link
- Birnkrant DJ, Panitch HB, Benditt JO, et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest. 2007;132(6):1977-86. PubMed
- American Academy of Pediatrics Section on C, Cardiac S. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005;116(6):1569-73. PubMed
- Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12(1):79. PubMed
- Beytia Mde L, Vry J, Kirschner J. Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives. Acta Myol. 2012;31(1):4-8. PubMed
- Mercuri E, Vilchez JJ, Boespflug-Tanguy O, et al. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2024;23(4):393-403. PubMed
- Morris P. Duchenne muscular dystrophy: a challenge for the anaesthetist. Paediatr Anaesth. 1997;7(1):1-4. PubMed
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.